Magnetyczny rezonans jądrowy (skrót NMR od ang. Nuclear Magnetic Rezonanse) jest zjawiskiem fizycznym, pozwalającym badać własności układu poddawanego działaniu fal z zakresu radiowego umieszczonego w stałym polu magnetycznym. W zjawisku NMR wykorzystywane są naturalne własności jąder atomowych i ich zachowanie w stałym i zmiennym polu magnetycznym. W zjawisku tym nie wykorzystuje się szkodliwego promieniowania, typu promieniowanie X, które jest nieobojętne dla materii, dlatego też metody pomiaru z wykorzystaniem NMR są zaliczane do nieinwazyjnych i nie zmieniających własności układu fizycznego. Metody NMR pozwalają badać strukturę i dynamikę badanego układu fizycznego od poziomu molekularnego do skali makroskopowej. Wykorzystywane są one szeroko w przemyśle m.in. w przemyśle spożywczym, chemicznym, farmaceutycznym oraz w diagnostyce medycznej. W artykule przedstawiono podstawy zjawiska NMR oraz opisano niektóre metody stosowane w badaniach jakościowych i ilościowych produktów spożywczych.
Słowa kluczowe: magnetyczny rezonans jądrowy, spektroskopia i relaksacja NMR, obrazowanie MR, TD NMR, SFC  Abstract
Abstract Nuclear Magnetic Resonance (NMR) is a physical phenomenon utilized to investigate molecular properties of matter by irradiating atomic nuclei in a magnetic field with radio waves. NMR uses natural properties of nuclei and their behaviour at static and radio frequency magnetic fields. NMR methods are non-destructive and non-invasive for matter. They allow to study structure and dynamics of physical systems from a molecular to macroscopic level. NMR methods are widely applied in food, chemical, pharmaceutical industries and in medical diagnostics. The basis of NMR and some its applications in quality control research of food products have been presented in the article.
Keywords: nuclear magnetic resonance, NMR spectroscopy and relaxation, magnetic resonance imaging (MRI), time domain NMR, SFC
WstępOdkrywcami zjawiska magnetycznego rezonansu jądrowego byli E.M. Purcell oraz F. Bloch, którzy opublikowali pierwsze prace z NMR-u w 1946 r. [1, 2]. Za swoje osiągnięcia otrzymali Nagrodę Nobla z fizyki w 1952 r. Pionierami badań NMR w Polsce byli już w latach pięćdziesiątych J. W. Hennel i A. Hrynkiewicz z Instytutu Fizyki Jądrowej w Krakowie [3]. Od momentu zarejestrowania pojedynczego sygnału magnetycznego rezonansu jądrowego w cieczy, w dziedzinie tej nastąpił ogromy postęp, po przez wielowymiarowa i wielokwantową spektroskopię fourierowską i tomografie rezonansu magnetycznego (MR). Twórcą spektroskopii fourierowskiej NMR (FT NMR) był Richard Ernst. Umożliwia ona obserwację widm NMR składających się z linii rezonansowych i analizę chemiczną materii. Za to osiągnięcie R. Ernst utytułowany został Nagrodą Nobla z chemii w 1966 r. Zastosowania dwuwymiarowej spektroskopii NMR w badaniach białek w biologii molekularnej przez Kurt’a Wüthrich’a również zostało nagrodzone Noblem z chemii w 2002 r. Ostatnia Nagroda Nobla z NMR-u została przyznana za obrazowanie magnetycznego rezonansu jądrowego (MR), a otrzymali ją Paul C. Lauterbur i Sir Peter Mansfield w 2003 r.
Różne metody NMR znalazły zastosowanie w badaniach materii do:- analizy chemicznej – po przez badanie struktury molekularnej i dynamiki,
- badań materiałów, które pozwalają określać własności fizyczne materii,
-obrazowania medycznego, czyli otrzymywania w sposób nieinwazyjny obrazów wnętrza ciała,
- inżynierii chemicznej – po przez pomiary dyfuzji, przepływów i rozkładu prędkości,
- badań w geofizyce i poszukiwaniach ropy – po przez rejestrację węglowodanów w skałach,
- badań w procesie kontroli jakości wykorzystując spektrometry niskiej rozdzielczości i przenośne sensory NMR.
Zasada zjawiska NMRZjawisko magnetycznego rezonansu jądrowego obserwujemy dla tzw. jąder magnetycznych. Jądra magnetyczne można wyobrazić sobie jako małe magnesiki, dipole magnetyczne, które mogą oddziaływać z otoczeniem. Wszystkie procesy, którym poddane są jądra atomowe zachodzą w mikroskopowym świecie i mogą być jedynie opisane przy pomocy abstrakcyjnej teorii matematycznej jaką jest mechanika kwantowa. Kryterium teorii mikroskopowej jest jej zgodność z założeniami teorii makroskopowej, przy przejściu z mikroświata do makroświata. W rzeczywistym eksperymencie magnetycznego rezonansu jądrowego mamy do czynienia z sygnałami otrzymywanymi z próbek makroskopowych. Dzięki istniejącej relacji pomiędzy teorią mikro i makroskopową zjawisko magnetycznego rezonansu jądrowego można opisać także na gruncie fizyki klasycznej.
Zjawisko magnetycznego rezonansu jądrowego w obrazie mikroskopowym[4, 5] Jądra atomowe można wyobrazić sobie także jako miniaturowe wirujące bąki, które opisuje się wielkością fizyczną, momentem pędu lub krętem . Z momentem pędu związany jest moment magnetyczny , który można wyrazić jako:

gdzie

jest czynnikiem giromagnetyczny, charakterystycznym dla danego jądra,

jest stałą Planka h podzielona przez 2

, jest oznaczeniem dla spinu jądrowego. Wielkość spinu jądrowego jest charakterystyczna i niezmienna dla danego jądra atomowego i jest wyrażona przez liczbę kwantową, która jest wielokrotnością liczby 1/2. Zjawisko magnetycznego rezonansu jądrowego obserwujemy tylko dla tych jąder, które maja niezerowy spin jądrowy, czyli charakteryzują się pewnym momentem magnetycznym. Są to jądra, które mają nieparzystą liczbę protonów albo neutronów. Do nich można zaliczyć jądra izotopów tych pierwiastków, które mają szczególne znaczenie do badania układów biologicznych jak np. jądra wodoru 1H. Poza jądrami wodoru, zwyczajowo nazywanymi protonami, jądrami magnetycznymi są np. jądra izotopu fosforu 31P, węgla 13C, azotu 15N czy fluoru 19F.
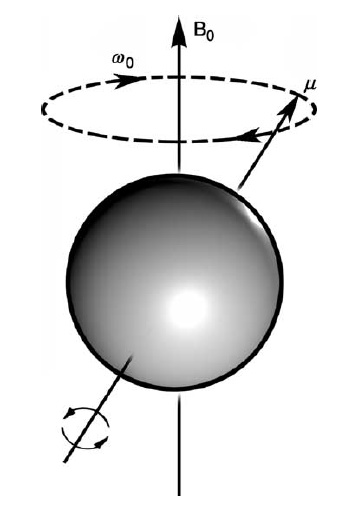
Rys.1. W zewnętrznym polu magnetycznym (B0) jądro atomowe precesuje z częstością  proporcjonalną do tego pola
proporcjonalną do tego pola  [6].
[6]. Opis zjawiska NMR można dalej przybliżyć do wirującego ruchu bąka mechanicznego w polu ciężkości Ziemi, który pod wpływem tego pola wykonuje dodatkowy ruch wokół tego pola. Ruch ten nazywa się precesją Larmora. Przez analogię za bąk magnetyczny można uznać jądro atomowe o danym spinie i momencie magnetycznym, umieszczone w stałym zewnętrznym polu magnetycznym o indukcji B0¬, w którym będzie wykonywać ruch wokół kierunku tego pola z częstością precesji Larmora
, która zależy od wielkości tego pola i rodzaju jądra, czyli od czynnika giromagnetycznego: 0 = B0 (Rys. 1). Jeśli na takie jądro, umieszczone w stałym polu magnetycznym B0, zadziałamy dodatkowym polem zmiennym B1, to w ogólnym przypadku niczego nie zaobserwujemy. Jeśli jednak częstość tego zmiennego pola B1, będzie równa częstości precesji Larmora

to zaobserwujemy silne oddziaływanie, które właśnie nazywane jest efektem rezonansu. Warunek rezonansu zapisuje się w postaci formuły matematycznej:
.
Zasadę magnetycznego rezonansu jądrowego w ujęciu kwantowym można także przedstawić w sposób obrazowy. Układ wielu małych magnesików jakimi są na przykład jądra wodoru w szklance wody, zachowuję się w nieobecności pola zewnętrznego w sposób nieuporządkowany. Jeśli umieścimy je w zewnętrznym polu magnetycznym następuje ich uporządkowanie, ale w sposób nieprzypadkowy. Dla jader o spinie I =1/2 ustawiają się one równolegle lub antyrównolegle do kierunku zewnętrznego pola magnetycznego, co odpowiada dwóm wartościom magnetycznej liczby kwantowej m = 1/2 oraz m = -1/2. Tym dwóm stanom odpowiadają dwa stany energetyczne (Rys. 2). Można indukować przejścia pomiędzy tymi stanami energetycznymi po przez dostarczenie energii promieniowania elektromagnetycznego (pola zmiennego wysokiej częstotliwości B¬1) , równej wartości różnicy energii poziomów energetycznych .
Dla jąder wodoru umieszczonych w polu magnetycznym B0 otrzymywanych obecnie w laboratoriach (od 1 do 23.5 Tesli) częstość rezonansu protonowego leży w zakresie od 42.58 MHz do 1 GHz, czyli w obszarze ultrakrótkich fal radiowych do mikrofal – zatem NMR jest działem radiospektroskopii.
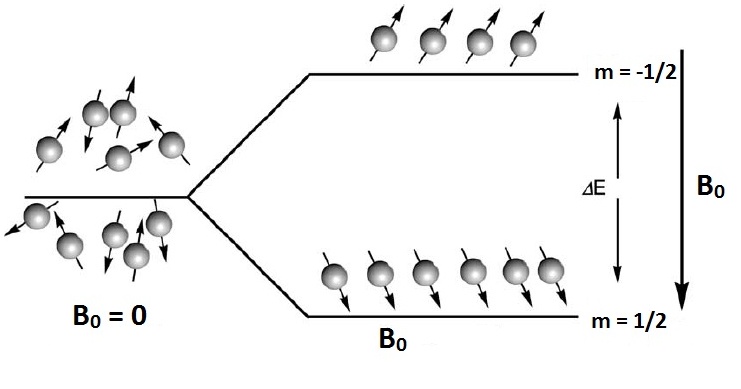
Rys. 2. Schematyczne przedstawienie jąder magnetycznych w nieobecności i w obecności stałego pola magnetycznego (rysunek zmodyfikowany z [6]). Obraz makroskopowy magnetycznego rezonansu jądrowego[4, 5] Liczba jąder w makroskopowej próbce jest ogromna, jeden centymetr sześcienny wody zawiera 1022 jąder wodoru, których momenty magnetyczne są całkowicie nieuporządkowane. W zewnętrznym polu magnetycznym B0 stosunek obsadzenia stanów równoległych N1/2 do stanów antyrównoległych N-1/2 opisuje prawo Boltzmanna:
,
gdzie k to stała Boltzmanna, T jest temperaturą w skali bezwzględnej. W temperaturze pokojowej i w polu B0 = 1 Tesli ten stosunek obsadzeń wynosi 10-6. Ten niewielki nadmiar spinów ustawionych równolegle do kierunku pola pozwala jednak na utworzenie mierzalnej makroskopowej magnetyzacji M0, która w stanie równowagi termodynamicznej jest ustawiona równolegle do kierunku pola B0 (Rys.3 A).
Zachowanie się magnetyzacji jest określone przez szereg procesów, z którymi mamy do czynienia w magnetycznym rezonansie jądrowym. Ruch składowych magnetyzacji w układzie xyz pod wpływem działania zaburzenia w postaci zmiennego pola magnetycznego B1, to proces relaksacji. Pole B1 w magnetycznym rezonansie jądrowym jest podawane w postaci krótko trwającym impulsów o określonej amplitudzie. Impulsy te maja tak dobrany czas trwania aby obróciły magnetyzację o określony kąt np. o kąt 90 stopni (Rys. 3 B). Po ustaniu działania takiego impulsu magnetyzacja wraca do stanu równowagi termodynamicznej, czyli relaksuje do stanu równowagowego. W magnetycznym rezonansie jądrowym oznacza się dwa procesy relaksacji i związane z nimi dwa czasy relaksacji: relaksacja spin-sieć i związany z nią czas T1 oraz relaksacja spin-spin i czas T2, które związane są z ruchem składowych magnetyzacji podłużnej i poprzecznej do pola B0¬ (Rys. 3 C i D). Wartości czasów relaksacji T1 i T2 wyznacza się eksperymentalnie i stanowią one ważny parametr opisujący materię, gdyż ich wartości zależą od otoczenia w jakim znajduje się jądra magnetyczne w próbce. Czasy relaksacji np. dla wody znajdującej się w różnym stanie skupienia będą się różnić o kilka rzędów wielkości.
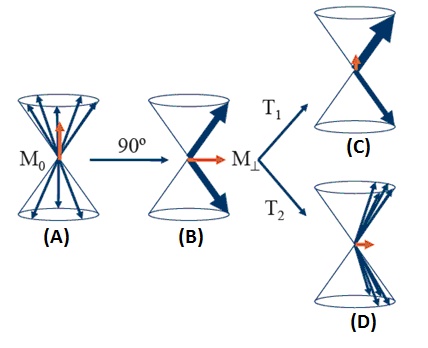
Rys. 3. Obrazowe przedstawienie zachowania się spinów jądrowych w prostym eksperymencie NMR (A) W stałym polu magnetycznym B0, spiny ustawiają się w położeniu równoległym i antyrównoległym do pola B0 zgodnie z rozkładem Boltzmanna¬, precesują przypadkowo wokół kierunku pola B¬0.¬ Magnetyzacja ma wartość maksymalną M0; (B) Po impulsie 90 pola B¬1 spiny precesują w fazie z częstością Larmora, magnetyzacja poprzeczna ma wartości maksymalną. (C) Relaksacja podłużna (spin-sieć). (D) Relaksacja poprzeczna (spin-spin). Rejestracja sygnału NMRAby zaobserwować sygnał NMR potrzebny jest spektrometr NMR, którego podstawowymi elementami jest magnes, wytwarzający silne i stałe w czasie pole B0, pomiędzy biegunami którego umieszczona jest w cewce próbka (Rys. 4 A). Cewka jest podłączona do nadajnika i odbiornika częstości radiowej, służącego do wytwarzania pola B1. Wszystkim steruje się przy pomocy komputera.
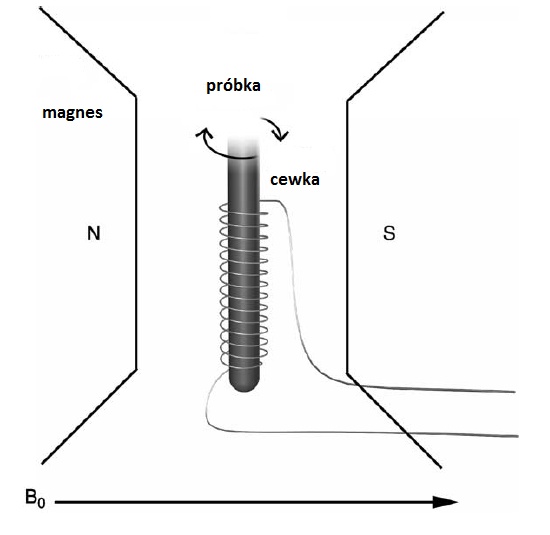
Rys. 4. (A) Schematyczne przedstawienie eksperymentu NMR. Próbka umieszczona jest w cewce, w której wytwarzane jest pole wysokie częstotliwości B¬¬1. Cewka umieszczona jest pomiędzy biegunami magnesu wytwarzającego stałe pole B0 (rysunek zmodyfikowany z [6]). (B) Spektrometr NMR w Laboratorium NMR, Zakładu Radiospektroskopii, Instytut Fizyki, Uniwersytet Jagielloński.Krótki impuls pola B1 wyprowadza magnetyzację z równowagi termodynamicznej. Po wyłączeniu tego pola magnetyzacja precesuje wokół pola B0 powodując modulacje pola magnetycznego co jest źródłem napięcia zmiennego powstałego w cewce. Zmiana napięcia w cewce rejestrowana jest w postaci sygnału zaniku swobodnej precesji (j. ang – Free Induction Decay, FID). Sygnał zaniku swobodnej precesji w czasie można poddać operacji matematycznej, zwanej transformatą Fouriera, która umożliwia obserwacje sygnału NMR w domenie częstotliwości w postaci widma. Spektroskopia NMR, szczególnie tzw. spektroskopia wysokiej zdolności rozdzielczej, jest oddzielną dziedziną NMR-u umożliwiającą analizę chemiczną układów, ale wymaga kosztownej aparatury, między innymi stosowania bardzo stabilnych pól magnetycznych z wykorzystaniem magnesów nadprzewodzących (Rys. 4B).
Szczególne zastosowania NMRMagnetyczny rezonans jądrowy jest upowszechniany dzięki zastosowaniom stosunkowo niedrogich i gabarytowo niewielkich spektrometrów, które można ustawić na stole w laboratorium („a benchtop spectrometer”). Urządzenia te wykorzystuje się do prostych eksperymentów magnetycznego rezonansu jądrowego w domenie czasu, często więc ta odmiana 1H NMR-u jest nazywana w skrócie TD-NMR („Time Domain NMR”) lub NMR-em niskiej rozdzielczości. Eksperymenty TD-NMR bazują na pomiarach relaksacyjnych i pomiarach dyfuzji, tzn. na różnicach w próbkach związanych z ich własnościami magnetyczno-rezonansowymi. Tego typu badania nie wymagają bardzo jednorodnych pól magnetycznych, technologia z wykorzystaniem magnesów trwałych jest tu wystarczająca, co czyni je stosunkowo tanimi i dostępnymi. Większość zastosowań TD-NMR korzysta z prostych impulsowych sekwencji pomiarowych, jak pomiaru sygnału zaniku swobodnej precesji (FID). Spektrometry TD-NMR są także wykorzystywane do pomiarów czasów relaksacji lub do przeprowadzania eksperymentów z użyciem impulsowych gradientów pola magnetycznego.
Własności magnetyczno-rezonansowe takie jak czasy relaksacji molekuł wody i innych substancji zależą silnie od otaczających ich innych molekuł. Generalnie, jądra wodoru tworzące fazę stałą, ciekłą jak woda czy olej dają sygnały NMR, które zanikają w różnym czasie. Woda krystaliczna, ciało stałe relaksuje w czasie kilkudziesięciu mikrosekund, natomiast woda związana charakteryzuje się pośrednim czasem relaksacji, sygnał NMR zanika w czasie kilkuset mikrosekund. Woda określana jako swobodna oraz substancje takie jak olej stanowiące składnik badanego materiału wyróżniają się czasem zaniku sygnału NMR od kilku milisekund do kilku sekund. Zastosowania TD-NMR bazują więc na obserwacji szybkości zaniku sygnału związanego z stopniem wiązania wody i stopniem swobody ruchów innych molekuł. Amplituda sygnału 1H NMR w domenie czasu odzwierciedla zawartość wodoru w różnych fazach. TD-NMR jest więc odpowiednią metodą do pomiarów jakościowych i ilościowych w laboratoriach i fabrykach, ponieważ oferuje szybkie analizy bez potrzeby trudnych przygotowań próbek. Metody TD-NMR obejmują zastosowania w wszystkich branżach przemysłowych poczynając od przemysłu związanego z produkcją żywności, po przez przemysły chemiczny, polimerowy i tekstylny do zastosowań medycznych i w branży farmaceutycznej. Zastosowania w produkcji żywności obejmują pomiary ilości wody i oleju w produktach spożywczych takich jak czekolada, produkty z kakao, w pieczywie i innych produktach zbożowych; pomiary zawartości tłuszczu stałego („solid fat content” – SFC) w margarynach i innych tłuszczach; analizę zawartości substancji oleistych i wody w nasionach, w mleku w proszku itp. W przemyśle chemicznym stosuje się metody TD-NMR do badań zawartości wody i oleju w proszkach czy zawartości wody w cemencie; w przemyśle polimerowym są to np. badania stopnia polimeryzacji i zawartości dodatków w polimerach i włóknach; w przemyśle tekstylnym badanie substancji kontrolujących stan włókien, na które składają się substancje oleiste, substancje powierzchniowo czynne i substancje antystatyczne. Przemysł paliwowy wykorzystuje pomiary TD-NMR do określania zawartości oleju w wosku i parafinie, zawartości wody w asfalcie, czy określania zawartości wodoru w węglowodorach. W branży medycznej wykorzystuję się pomiary TD-NMR do badań środków kontrastujących stosowanych w obrazowaniu MR, w przemyśle farmaceutycznym bada się np. zawartość wody i oleju w tabletkach i proszkach [7, 8].
Wyznaczanie zawartości tłuszczu stałego (SFC) Pomiar zawartości tłuszczu stałego (w fazie stałej) w jadalnych olejach i tłuszczach jest niezbędnym badaniem wykorzystanym w przemyśle piekarniczym, cukierniczym i przy produkcji margaryny. Tłuszcze i oleje są zwykle mieszaniną wielu związków lipidowych, charakteryzujących się pewnym zakresem topnienia. Zakres topnienia wielu lipidów żywnościowych pojawia się w temperaturach używanych w przygotowywaniu żywności, przechowywaniu i ich innym wykorzystaniu. Ponieważ stosunek tłuszczu stałego do tłuszczu w fazie ciekłej wpływa na strukturę, stabilność i smak jedzenia, zakres topnienia jest niezwykle istotnym parametrem w procesie kontroli jakości.
Tradycyjna metoda używana do określania zawartości tłuszczów stałych bazuje na pomiarze rozszerzalności cieplnej przy pomocy dylatometru [9]. Metoda ta polega na pomiarze zmiany objętości stałej ilości próbki w skutek podwyższenia temperatury. Stopień rozszerzalności jest związany z zmianą w zawartości frakcji stałociałowej, która pozwala na określenie współczynnika tłuszczu stałego („solid fat index - SFI”). Mimo, że metodę tą uważa się obecnie jako wolną, niedokładną i nieefektywną, pomiar SFI pozostaje nadal metodą rekomendowaną przez AOCS [10]. W dużym stopniu jest jednak zastępowana przez metodę bazująca na pomiarach TD-NMR.
Metoda pomiaru zawartości tłuszczu stałego (SFC) wykorzystująca właśnie zjawisko magnetycznego rezonansu jądrowego w domenie czasu została zaadaptowana w przemyśle i ustanowiono dla niej międzynarodowe standardy takie jak: AOCS CD16B-93 [11, 12] oraz ISO 8292 [13] i IUPAC 2.150 [14] (dla Europy) [10]. Jedną z zalet metody z wykorzystaniem zjawiska NMR jest szybkość pomiaru (zwykle kilkanaście sekund na jeden pomiar). Ponadto metoda jest nie destruktywna, więc można wygodnie powtarzać pomiary bez niszczenia próbki. Pomiar SFC może być wykonywany, po uprzednim przeszkoleniu, przez osoby nie będące specjalistami w zakresie magnetycznego rezonansu jądrowego.
Zawartość tłuszczu stałego jest wyznaczana po przez rejestrację sygnału NMR w domenie czasu pochodzącego zarówno od cieczowej i stałociałowej komponenty w próbce tłuszczu (metoda bezpośrednia), lub po przez wykrywanie zmian w sygnale pochodzącym od cieczy gdy jest zamieniany przez sygnał pochodzący od frakcji stałociałowej (metoda pośrednia) [7, 10, 15]. W większości nowoczesnych spektrometrów wykorzystywanych w tego typu pomiarach stosuje się metodę impulsową w eksperymencie NMR. Spektrometr rejestruje ewolucję sygnału NMR, zawierającego analityczną informację o frakcjach wodoru w stanie ciekłym i stałym. Po impulsie sygnał jest rejestrowany w zakresie od mikrosekund do sekund. Zanik sygnału NMR wynika z procesu relaksacji. Początkowy, zanikający gwałtownie sygnał pochodzi od frakcji stałociałowej, która relaksuje szybko do stanu równowagi (a), pozostała część sygnału, zanikająca wolniej, pochodząca od frakcji cieczowej, charakteryzuje się wolniejszą relaksacją (b) (Rys. 5).
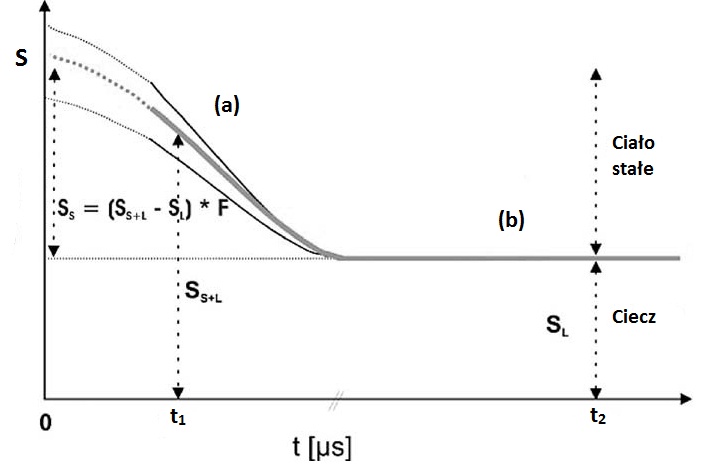
Rys. 5. Sygnał FID (S) w funkcji czasu t i jego analiza dla wyznaczenia SFC. Początkowa, wykropkowana część FID-u to obszar tzw. czasu martwego spektrometru (rysunek zmodyfikowany z [8]).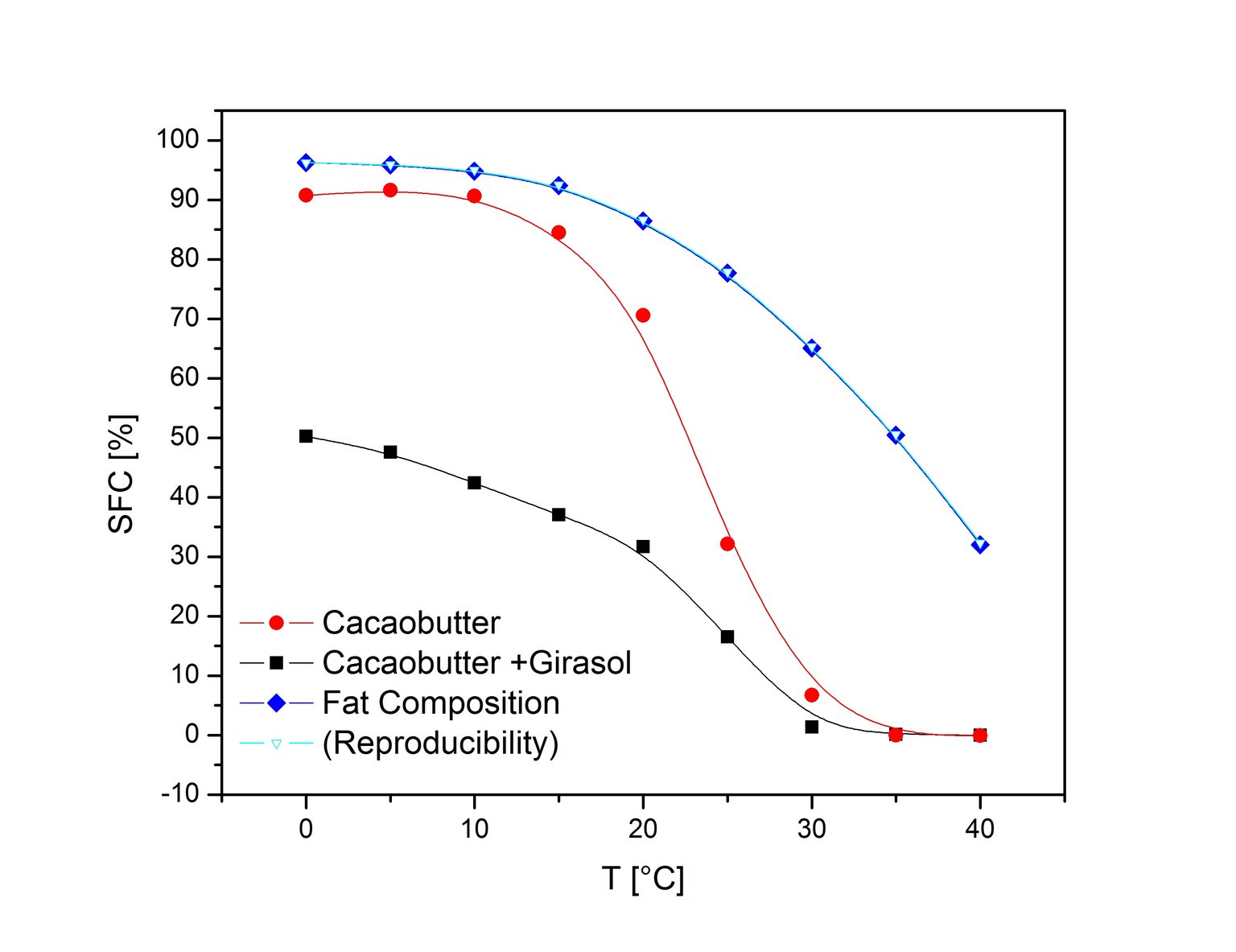
Rys. 6 Zależność SFC od temperatury dla kilku produktów [7, 8]. Metoda bezpośrednia SFCMetoda bezpośrednia jest szybka, powtarzalna, a przygotowanie próbki jest minimalne. Stopiony olej jest wlewany bezpośrednio do próbówki NMR, w której jest poddawany procesowi utwardzania. W procesie utwardzania próbka poddawana jest działaniu określonej temperatury przez określony czas, procedura ta jest także ustandaryzowana (IUPAC, AOCS). Tylko jeden pomiar NMR jest potrzebny aby otrzymać wartość SFC w metodzie bezpośredniej. Aby otrzymać wykres topnienia należy przeprowadzić takie pojedyncze pomiary w kilku temperaturach (Rys. 6). Jednakże odtwarzanie całego profilu topnienia dla danej próbki nie zawsze jest konieczne. Kiedy mierzy się zawartość tłuszczu stałego produktu na pewnym etapie produkcji lub porównuje się produkty podczas przechowywania, wtedy wystarczy jeden pomiar NMR w stałej temperaturze.
W metodzie bezpośredniej mierzy się wartość amplitudy w dwóch punktach na sygnale FID, które pozwalają wyznaczyć wartość amplitudy dla fazy cieczowej+ stałociałowa (SS+L) dla czasu t1 (Rys. 5 ) i odczytuje się wartość sygnału dla frakcji cieczowej w punkcie t2 (SL). Następnie z prostej zależności wyznacza się wartość SFC:

gdzie F jest czynnikiem poprawiającym dokładność pomiaru, umożliwiającym oszacowanie sygnału zaraz po ustaniu impulsu, co jest niemożliwe do rejestracji z powodu tzw. czasu martwego spektrometru; D jest tzw. czynnikiem przesunięcia cyfrowego, ustalanym na drodze pomiarów kalibracyjnych. Czynnik F wyznacza się także z pomiarów kalibracyjnych przy pomocy standardowych substancji zwykle mających SFC równe 0 %, 30 % i 70 %, i dostarczanych przez producenta spektrometru.
Metoda pośrednia SFC Metoda pośrednia wymaga wykonania czterech pomiarów w dwóch temperaturach aby obliczyć zawartość frakcji stałej tłuszczu [15]. Wykorzystuje się próbkę ciekłego oleju jako próbkę standardową, która podobnie jak badana próbka jest mierzona w dwóch temperaturach. Użycie ciekłego oleju jako próbki standardowej bierze pod uwagę zmianę czułość instrumentu wraz ze zmiana temperatury. W metodzie tej odczytuje się wartość amplitudy sygnału cieczowego SL w jednym punkcie (t2) (Rys. 5) w danej temperaturze T oraz w temperaturze gdy cała próbka jest już stopiona (T¬m), zwykle jest to 60C. Wartość SFC wylicza się z zależności:

gdzie SLst dotyczy pomiarów amplitudy dla próbki standardowej.
Mimo, że metody pomiaru SFC zostały ustandaryzowane, istnieją pewne ograniczenia tych procedur. Słabością stosowania współczynnika F w pomiarze bezpośrednim jest na przykład fakt, że przyjmuję się jego wartość dla wszystkich mierzonych próbek, pomijając zależność od wartości czasów relaksacji, temperatury oraz obecności form krystalicznych jakie mogą być tworzone i odzwierciedlone w kształcie otrzymanego sygnału FID. Metoda pośrednia daje natomiast duże wartości niepewności standardowych w porównaniu do metody bezpośredniej. Dlatego też, stale poszukuje się innych rozwiązań w metodach NMR, które pomogłyby w sposób efektywny badać jakość produktów spożywczych[16].
Autor: Magdalena WitekLiteratura:1. Bloch F., Hansen W. W. and Packard M., Nuclear Induction. Physical Review, 1946. 69(3-4): p. 127-127.
2. Purcell E. M., Torrey H. C. and Pound R. V., Resonance Absorption by Nuclear Magnetic Moments in a Solid. Physical Review, 1946. 69(1-2): p. 37-38.
3. J. W. Hennel, A.Z.H., Arch. Sci. (Genawa), 1958. 11.
4. Hennel J. W and Klinowski J., Podstawy magnetycznego rezonansu jądrowego. . 2000, Poznań: Wydawnictwo Naukowe UAM.
5. Stankowski J. and Hilczer W., Pierwszy krok ku radiospektroskopii rezonansów magnetycznych. 1994, Poznań: OWN.
6. Schmidt, S.J., Water and Solids Mobility in Foods, in Advances in Food and Nutrition Research. 2004, Academic Press. p. 1-101.
7. http://www.brukeroptics.com/solidfat.html. [cited.
8. Todt, H., G. Guthausen, W. Burk, D. Schmalbein, et al., Time-Domain NMR in Quality Control: Standard Applications in Food Modern Magnetic Resonance. 2006, Springer Netherlands. p. 1739-1743.
9. Hannewijk J., Haighton A. and Hendrikse P. W., Dilatometry of fats, in Analysis and Characterisation of Oils, Fats and Fat Products, B.H. A., Editor. 1964, Interscience Publishers: London. p. 119-182.
10. Firestone D., Official Methods and Recommended Practices of the AOCS. American Oil Chemists' Society, Champaign, I11. 1998.
11. AOCS Official Method Cd 16b - Solid fat content (SFC) by low resolution magnetic resonance. 1993, American Oil Chemist Society.
12. Bruker minispec: Food Industry and Application. 1970. Notes Numbers 1, 2.
13. Animal and vegetable fats and oils - Determination of soild fat content - Pulsed nuclear magnetic resonance method. 1991, ISO 8292 International Standard.
14. Soild content determination in fat by NMR - Low resolution nuclear magnetic resonance. 1987, IUPAC Norm Version 2.150 (ex. 2.323).
15. Coupland J., Determination of solid fat content by nuclear magnetic resonance. Current Protocols in Food Analytical Chemistry, 2001: p. D3.1.1-D3.1.8.
16. van Duynhoven J., Voda A., Witek M. and Van As H., Chapter 3 - Time-Domain NMR Applied to Food Products, in Annual Reports on NMR Spectroscopy, Webb G. A., Editor. 2010, Academic Press. p. 145-197.










Recenzje