Streszczenie
W sferze dostępnych technologii pomiarowych magnetyczny rezonans jądrowy (NMR) zajmuje wyjątkową pozycję ze względu na szeroki zakres zastosowań, szczególnie w przemyśle spożywczym. Metody NMR mogą być stosowane w różnych obszarach umożliwiając badanie mezo- i mikrostruktury układów, skład produktów, a także wpływ fizjologiczny i psychologiczny na konsumenta. W artykule przedstawiono kontynuacje zastosowań metod NMR w badaniach jakościowych i ilościowych produktów spożywczych, w szczególności zastosowań spektroskopii NMR.
Spektroskopia NMR
Spektroskopia NMR, a szczególnie spektroskopia wysokiej zdolności rozdzielczej, jest dziedziną NMR-u umożliwiającą analizę składu chemicznego układów. Spektrometr do tego typu pomiarów jest wyposażony w magnes nadprzewodzący, wytwarzający silne i jednorodne pole magnetyczne B0, a próbki badawcze są zazwyczaj w postaci ciekłych roztworów danej substancji w rozpuszczalniku pozbawionym jąder rezonansowych (przykładowo dla rezonansu protonowego rozpuszczalnik jest całkowicie zdeuterowany). Pośród wszystkich metod spektroskopowych, NMR jest jedyną metodą, od której oczekuje się całkowitej analizy i interpretacji widma. Mimo, że większa ilość substancji potrzebna jest do badania w porównaniu do metody spektroskopii masowej (MS), spektroskopia NMR nie jest destruktywna dla próbki, a wykorzystując nowoczesne urządzenia można otrzymać widma dobrej jakości dla próbek o masie poniżej miligrama [1, 2].
Widma NMR można zarejestrować dla jąder rezonansowych różnych pierwiastków, przykładowo dla jąder wodoru 1H, czyli dla protonów, jąder 13C czy jąder 13P. Momenty magnetyczne () dla różnych jąder rezonansowych mają różne wartości, a to powoduje, że w danym stałym polu magnetycznym B0 mają różne wartości częstotliwości rezonansowej ( ). Ponieważ wszystkie jądra atomowe jednego rodzaju (dla danego pierwiastka) maja taki sam moment magnetyczny, oczekuje się, że będą dawać sygnał rezonansowy dla tej samej wartości częstotliwości. Jednakże w stałym polu magnetycznym dla danego związku chemicznego możemy obserwować więcej niż jedną linię rezonansową, dla jąder w cząsteczce rezonans zachodzi dla trochę różnych wartości pola magnetycznego. Jest to spowodowane obecnością w otoczeniu jądra atomowego elektronów i jonów, które generują w polu B0 dodatkowe pole magnetyczne. To dodatkowe pole magnetyczne ekranuje jądra od zastosowanego pola B0 tak, że pole w miejscu obserwowanego jądra jest nieco inne niż pole zewnętrzne B0. Wielkość tego dodatkowego pola jest bardzo mała w porównaniu do pola B0 i zakresy jego działania określa się w jednostkach ppm, czyli w częściach na milion (parts per milion). Dla większości związków organicznych linie rezonansowe dla jąder wodoru mieszczą się w zakresie 12 ppm, dlatego też w rezonansie protonowym wykorzystuje się bardzo precyzyjne spektrometry aby rozdzielić strukturalnie różne atomy wodoru pośród tak wąskiego zakresu [1].
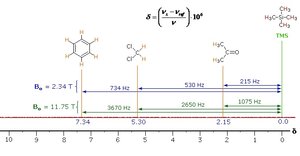
W przeciwieństwie do spektroskopii UV czy w podczerwieni, gdzie linie absorpcyjne są unikalnie zlokalizowane przez częstotliwość czy długość fali, umiejscowienie różnych sygnałów rezonansowych NMR zależy zarówno od siły pola magnetycznego B0 jak i od częstości pola radiowego. Dlatego też wykorzystuje się sygnały referencyjne pochodzące od substancji standardowych dodawanych do badanej próbki i do położenia sygnału referencyjnego odnosi się pozostałe sygnały w widmie. Taka standardowa substancja powinna być obojętna chemiczne i dawać pojedynczą linię rezonansową w widmie NMR. Przykładowo dla rezonansu wodorowego często wykorzystuje się tetrametylosilan (CH3¬)4Si (TMS). Różnica w częstotliwości pomiędzy sygnałem pochodzącym od próbki lub danej linii (s) i sygnałem referencyjnym (ref), podzielona przez częstość podstawową spektrometru (¬) i pomnożona przez milion daje wartość w ppm i określana jest jako przesunięcie chemiczne () (Rys. 1.) Dla tak przyjętych jednostek odległości linii nie zależą od częstości rezonansowej spektrometru.
Natężenie pola magnetycznego B0 istotnie wpływa na zdolność rozdzielczą spektrometru, im jest ono większe tym więcej linii rozdziela się w jednej jednostce ppm.
Rys. 1 Ilustracja przesunięcia chemicznego obliczanego względem substancji standardowej TMS dla acetonu, chlorku metylu i benzenu [1].
Amplituda lub intensywność sygnałów rezonansowych NMR, proporcjonalna jest do molarnej koncentracji próbki. Dlatego też małe ilości lub próbki bardzo rozpuszczone będą dawać słabe sygnały, podwojenie lub potrojenie koncentracji próbki zwiększy proporcjonalnie siłę sygnału rezonansowego. Jeśli weźmiemy natomiast tą samą ilość molarną dwóch różnych związków, ale różniących się ilością jąder rezonansowych na cząsteczkę, to otrzymane sygnały rezonansowe będą charakteryzować się większą intensywnością dla tej substancji, która ma więcej jąder rezonansowych w cząsteczce. Jest to ważne w przypadku próbek składających się dwóch lub większej ilości grup jąder rezonansowych (przykładowo dla alkoholu etylowego mamy grupy protonowe OH, CH2, CH3), gdyż umożliwia to określenie wzajemnego stosunku atomów jąder rezonansowych w każdym z różnych grup wodorowych (Rys. 2). Kolejną obserwacja w widmach NMR jest rozszczepianie sygnałów związków mających różne grupy atomów wodoru związane do sąsiadujących atomów węgla (przykładowo jak dla alkoholu etylowego na Rys. 2). Jest to związane z oddziaływaniem zwanym sprzężeniem spinowo-spinowym lub sprzężeniem J. Oddziaływanie J zachodzi za pośrednictwem wiązań chemicznych molekuły. Rozczepienie sygnału spowodowane tym oddziaływaniem jest zwykle małe, rzędu Hz i jest oznaczane jako stała wiązania J. Takie rozszczepienia tworzą dublety, triplety i są łatwo rozpoznawalne w widmie, gdyż są symetrycznie rozlokowane wokół linii centralnej. Innym oddziaływaniem mającym wpływ na widmo NMR jest oddziaływanie dipolowe, które jest oddziaływaniem dipoli magnetycznych, którymi są jądra rezonansowe. Oddziaływanie dipolowe wpływa na poszerzenie linii widmowych. Spektroskopia NMR o dużej zdolności rozdzielczej pozwala więc scharakteryzować otoczenie chemiczne badanego jądra [1, 2].
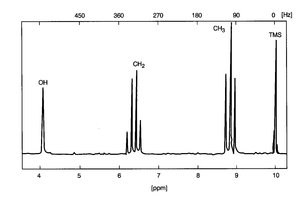
Rys. 2 Widmo NMR alkoholu etylowego. Pola pod liniami OH, CH2, CH3 maja się do siebie jak 43:80:155, co daje w przybliżeniu stosunek intensywności 1:2:3 [2].
Widma protonowe różnych próbek biologicznych (np. białek) składają się z bardzo dużej ilości, nakładających się na siebie linii widmowych, dlatego też często stosuję się techniki NMR umożliwiające pomiar widm wielowymiarowych. Przykładami metod dwuwymiarowych, szczególnie przydatnych w badaniach substancji biologicznych są: metoda COSY (ang. Corelation Spectroscopy) oraz NOESY (ang. Nuclear Overhauser Effect Spectroscopy). Istotą tych metod jest zastosowanie sekwencji dwóch (COSY) lub trzech (NOESY) impulsów pola zmiennego oddzielonych od siebie określonymi czasami. Proces otrzymywania widm w spektroskopii dwuwymiarowej można podzielić więc na kilka etapów: przygotowanie, ewolucję, mieszanie (NOESY) oraz detekcję. Po pierwszym impulsie przygotowawczym jądra rezonansowe oddziałują ze sobą, co przejawia się wzajemną modulacją ich częstości precesji, po drugim impulsie, w czasie detekcji sygnału FID, otrzymuje się informacje o procesach relaksacji, o sprzężeniach i wzajemnych oddziaływaniach zachodzących w czasie między impulsami. Informacje te zostają pokazane po przeprowadzeniu dwuwymiarowej transformaty Fouriera (2D FT). W dwuwymiarowym widmie będącym wynikiem 2D FT obserwuje się obok widma jednowymiarowego położonego na przekątnej, także linie poza przekątną (tzw. cross peaks), które informują o wzajemnych sprzężeniach między protonami. Dzięki temu możliwa jest identyfikacja i przyporządkowanie linii do odpowiednich grup protonowych w cząsteczce. W metodzie NOESY intensywności linii rezonansowych związane są z tzw. przejściami cross-relaksacyjnymi, które są wywołane oddziaływaniami dipolowymi w poszczególnych parach jąder. Metoda ta pozwala na wyznaczenie odległości proton-proton oraz umiejscowienia protonów w skomplikowanej molekule ([2] i literatura zawarta w tym opracowaniu).
Zastosowanie spektroskopii wysokiej zdolności rozdzielczej NMR
Jedną z możliwości zastosowania spektroskopii wysokiej zdolności NMR jest jej przydatność w dziedzinie nauki niezwykle popularnej przez ostatnie lata, a określanej jako metabonomika lub metabolomika. Metabonomikę definiuje się jako ilościowe pomiary odpowiedzi metabolicznej opisanej przez wiele parametrów żywego układu na patofizjologiczne bodźce lub modyfikacje genetyczne [3]. Pojęcie metabolomiki, zostało wymyślone jakiś czas po stworzeniu metabonomiki i istnieje dla niego bardzo wiele definicji. Przykładowo jest ono opisywane jako: systematyczne badania unikatowych znaczników chemicznych, które pozostają po specyficznych procesach zachodzących na poziomie komórkowym lub badanie zestawu wszystkich metabolitów o niskiej wadze molekularnej powstałych pod wpływem danych czynników [4].
Skład metabolitów w układach biologicznych jest zwykle złożoną mieszaniną wielu związków organicznych i nieorganicznych soli. Analiza złożonych mieszanin wymaga znaczących możliwości analitycznych ze strony istniejących technologii. Istnieje kilka technologii analitycznych, a są nimi metody [5] chromatograficzne (chromatografia gazowa - GS i cieczowa – HPLC, UPLC), spektroskopia masowa (MS) oraz metody NMR. Chromatografia i MS są dobrze rozwiniętymi selektywnymi metodami detekcji, oferującymi czułość detekcji, która zmienia się w zależności od zawartości metabolitu [5]. W metodach chromatograficznych mierzone jest oddziaływanie pomiędzy analitem i materiałem stacjonarnym, natomiast informacja strukturalna nie jest bezpośrednio otrzymywana. MS umożliwia pomiar masy metabolitów i ich fragmentów, daje więc informacje o formule metabolitów, ale niedostatecznie umożliwia identyfikację swoistych molekuł, które są ważne w kontekście układów biologicznych [5]. Kombinacja obu metod daje pewne ulepszenia, ale jednoznaczna detekcja metabolitów i określenie struktury z wykorzystaniem obu metod wymaga dostępności określonych standardów; określenie ilościowe metabolitów z użyciem obu metod wymaga procedur kalibracyjnych z wykorzystaniem właśnie określonych związków. Ponadto, metody te wymagają intensywnych ekstrakcji chemicznych i czasami reakcji chemicznych, co prowadzi do całkowitej utraty informacji o układzie in vivo.
W przeciwieństwie do obu opisanych metod, metody NMR dostarczają informacji o połączeniach atomowych w molekułach w czystej postaci lub w mieszaninach i oferują uniwersalną detekcje ilościową wszystkich metabolitów. Jako metoda nie destruktywna pozwala na badania in vivo i jest skutecznym rozwiązaniem do bezpośrednich badań nienaruszonych tkanek z wykorzystaniem pomiarów NMR wysokiej zdolności rozdzielczej i wirowania pod kątem magicznym. Z pomiarów NMR można też otrzymać informacje o dynamice molekularnej i oddziaływaniach. Mała czułość metody NMR w porównaniu do standardowych metod (chromatografii i MS) pozostaje pewnym problemem, jednakże można ją zwiększyć dzięki zastosowaniu technologii sond kriogenicznych wykorzystywanych w spektrometrach NMR. Stosowanie sond kriogenicznych znacznie skraca czas pomiarów i możliwość otrzymania informacji o składzie metabolitów. Mimo, że aparatura NMR jest ogólnie bardzo droga, wydajność w sensie określania struktury, wykrywania nowych biomarkerów, informacji o składzie czyni NMR tańszym, kiedy liczymy koszt na jedną próbkę (~10 USD na próbkę) [6]. Zastosowanie spektroskopii NMR w matabonomice wymaga również wykorzystania odpowiednich wielowymiarowych analiz matematycznych otrzymanych danych, przykładowo metody analizy głównych składowych (ang. Principal Component Analysis, PCA), która jest metodą używaną do zmniejszania rozmiaru danych statystycznych, w przypadku gdy mamy zbiory danych składających z wielu obserwacji, których każda składa się z wielu zmiennych [7, 8].
Oczywistym jest, że żadna z znanych technik samodzielnie nie jest w stanie rozwiązać wszystkich problemów napotykanych w analizie metabolitów. Wybór metody jest uzależniony od celu badań. Kombinacja wszystkich metod (LC-NMR-MS) [9] jest w wielu przypadkach najprawdopodobniej najlepszym rozwiązaniem.
Spektroskopia NMR i matabonomika w kontroli jakości żywności
Przedmiotem metabonomiki/metabolomiki jest także badanie płynnych produktów żywnościowych dla celów kontroli jakości. Kontrola jakości bazuje tutaj na składzie chemicznym żywności, który określany jest w skali globalnej. Metody spektroskopowe NMR znalazły tutaj szczególe zastosowanie w badaniach jakościowych napojów oraz innych płynnych produktach żywnościowych. Jednym z pierwszych przykładów, było zastosowanie spektroskopii NMR do badania składu soków owocowych oraz octów winnych [10]; gdzie udało się oznaczyć wiele z chemicznych składników i udowodniono możliwość wykorzystania spektroskopii wysokiej zdolność rozdzielczej do badań ciekłych produktów żywnościowych. W zestawieniu z wielowymiarową analizą danych, spektroskopia NMR umożliwiła rozróżnianie soków pochodzących z różnych odmian jabłek [11, 12]. Wyniki tych pomiarów pokazały duży potencjał metody do identyfikacji soków jabłkowych ze względu na pochodzenie, ale także do badania zmian biochemicznych zachodzących w samych owocach. Identyfikacja kilkudziesięciu sygnałów w widmach NMR wraz z wielowymiarową analizą danych, umożliwiła także zastosowania spektroskopii NMR do wykrywania fałszowania soków pomarańczowych [13].
Zastosowanie spektroskopii 1H NMR wraz z analizą głównych składowych (PCA) wykorzystano także w badaniach 191 odmian zielonej herbaty pochodzących z różnych krajów [14]. Przykładowo, analiza PCA pokazała rozdzielenie pomiędzy chińska herbatą Longjing a innymi rodzajami herbaty pochodzącymi z Chin. Zarejestrowano kilka markerów metabolicznych, np. herbata Longjing zawierała wysoką zawartość teaniny, kwasu galusowego, kofeiny, galusanu epigallokatechiny, galusanu epikatechiny i niski poziom epigallokatechiny w porównaniu do innych herbat. Pozwoliło to na ustanowienie pewnych znaczników w przypadku badania dużej ilości herbaty nieznanego pochodzenia i składu. Inne badania pokazały potencjał kombinacji NMR-u i chemometrii w kontroli jakości herbaty rumiankowej ze względu na jej pochodzenie, czystość i metody modyfikowania [15]. Metody spektroskopowe NMR wykorzystano także w procesie kontroli jakości przy produkcji piwa i badaniu jego autentyczności [16].
Klasyfikowanie soków owocowych przy pomocy NMR (SGF Profiling)
Kontrola jakości jest ciągłym wyzwaniem w przemyśle spożywczym w związku z oszustwami takimi jak złe oznakowanie produktu lub typu i pochodzenia jego składników. Dla producentów soków owocowych istotna jest także ochrona dobrego wizerunku ich produktów oraz ochrona przed nieuczciwymi producentami. Spektroskopia NMR w połączeniu z zastosowaniem wieloparametrowej analizy statystycznej jest niezwykle uniwersalnym narzędziem umożliwiającym aby podjąć to wyzwanie. Jednym z udanych zastosowań jest właśnie technika klasyfikowania soków owocowych (Bruker JuiceScreenerTM for SGF ProfilingTM [17, 18]). Metoda ta powstała dzięki współpracy producenta spektrometrów firmy Bruker BioSpin GmbH oraz SGF International e.V. (SURE-GLOBAL-FAIR), stowarzyszenia przemysłowców, mającego na celu samokontrolę i walkę o przestrzeganie standardów jakości i bezpieczeństwa oraz bezpieczny i uczciwy światowy rynek soków owocowych w imieniu jej członków i konsumentów. SGF jest członkiem stowarzyszenia EQCS (European Quality Control System) i jej oddział SGF/IRMA (International Raw Material Assurance) kontroluje rynek producentów soków w ponad 50 krajach świata.
Zaproponowany przez firmę Bruker system jest w zautomatyzowany, łącznie z podajnikiem próbek i samą procedurą stabilizacji, przygotowywania do eksperymentu, pomiarem, analizą danych i raportowaniem. Przygotowanie każdej z próbki jest minimalne, potrzeba dodać tylko około 10 % bufora składającego się z D2O (ciężkiej wody). Zwykle przeprowadza się dwa eksperymenty NMR: wersję eksperymentu z wykorzystaniem jednowymiarowej sekwencji pomiarowej 1D-NOESY, pozwalającej na ilościowe oszacowanie składników mieszaniny odznaczających się sygnałami w widmie NMR blisko położenia sygnału od wody; oraz szybką sekwencję dwuwymiarową 2D z analizą sprzężeń J, która ułatwia jednoznaczną identyfikację sygnałów. Dla każdej próbki soku owocowego określa się parametry zwiane z jakością i autentycznością, wykorzystując procedury pomiarowe, które łącznie z końcowymi wynikami trwają tylko kilkanaście minut [19].
Metoda klasyfikacji soków owocowych charakteryzuje się niskim kosztem na próbkę i jest konkurencyjna w stosunku do konwencjonalnych metod kontroli jakości. Obecnie baza zawiera widma ponad 6 000 próbek soków referencyjnych, w tym obejmuje 1500 autentycznych certyfikowanych próbek pobranych przez inspektorów SGF z różnych obszarów upraw oraz dla różnych odmian owoców.
Analiza widm NMR odbywa się w dwóch etapach. W pierwszym etapie identyfikuje się i oszacowuje ilościowo indywidualne składniki, natomiast w drugim kroku stosuje się metody statystyczne do wykrywania nieoczekiwanych odstępstw lub określania pochodzenia próbki, składu owoców lub dodawania innych typów owoców.
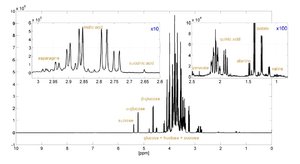
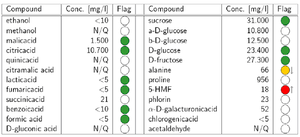
W porównaniu do próbek referencyjnych, specyficzne odstępstwa w koncentracji konkretnych związków lub w kombinacji tych związków mogą wskazywać na określony problem w jakości i autentyczności. Informacja w widmie NMR umożliwia wyszczególnienie wielu związków jednocześnie (Rys. 3) i wyznaczenie koncentracji dla kilkudziesięciu charakterystycznych związków występujących sokach (Tabela 1). Umożliwia także detekcje oszustw jak dodawanie cukru, kwasu cytrynowego lub soku cytrynowego (przykładowo w sokach jabłkowych) czy używanie niedobranych owoców (odznaczająca się wysoką koncentracją kwasu chinowego w soku jabłkowym).
Rys. 3 Widmo 1H NMR soku jabłkowego z powiększeniem dziesięciokrotnym i stukrotnym dwóch obszarów pokazujących sygnały od najistotniejsze związków [19].
Tabela 1. Oszacowanie zawartości składników soku pomarańczowego. Kolorowe oznaczenia pokazują stopień zgodności lub odstępstwa od zakresów referencyjnych, dostarczonych przez A.I.J.N (European Fruit Juice Association); (N/Q – oznacza brak danych z powodu zbyt małej intensywności sygnału lub niedostatecznej identyfikacji linii rezonansowej) [19].
W drugim kroku metody SGF Profiling wykorzystuje się analizę statystyczną opierającą się na bazie widm referencyjnych. Stosuje się tu kilka podejść, które obejmują testy statystyczne i klasyfikację oraz rozróżnianie dla wielu aspektów jakościowych. Testy i modele statystyczne mogą być zastosowane do pojedynczego widma, i służą uzyskaniu określonej informacji dotyczącej: separacji typu owocu z którego wyprodukowano sok, rozróżnieniu pomiędzy odmianami owoców cytrusowych, rozróżnieniu pomiędzy kategorią produktu (przykładowo sok pomarańczowy i sok pomarańczowy otrzymany z koncentratu) lub charakteryzowaniu różnic w składzie dla dwóch grup podobnych produktów (np. koncentratów soku jabłkowego pochodzących z Polski i z Chin) [19].
Zwykle dla danej próbki w pierwszym podejściu oszacowuje się rodzaj owoców z których wyprodukowano dany sok. Model jest w stanie rozróżnić pomiędzy 20 rodzajami owoców. Umożliwia to przykładowo rozpoznanie dodawania soku z mandarynki do soku pomarańczowego. Sok z mandarynek jest tańszy i często producenci dodają go bez zamieszczenia tej informacji (co jest niedozwolone w Europie). Metody spektroskopowe NMR są w stanie wykryć zawartość soku z mandarynek na poziome około 10 %. Wykorzystując bardziej wyspecjalizowane modele, można w stosunku do soków z pomarańczy i jabłek rozróżnić pomiędzy sokiem uzyskanym bezpośrednio i sokiem z koncentratu oraz odnotować pochodzenie owoców. Informacje opracowywane są przez określony program i przedstawiane w sposób graficzny. Rys. 4 pokazuje przykładowe wyniki analizy dla soków pomarańczowego i jabłkowego, wyróżniające kraj pochodzenie soku. Pochodzenie geograficzne soku owocowego jest istotnym aspektem komercyjnym. Trójwymiarowe projekcje wykorzystanego modelu w przestrzeni pokazują elipsoidy prawdopodobieństwa dla każdego z wyszczególnionych źródeł, badana próbka oznaczona jest czerwoną gwiazdką.
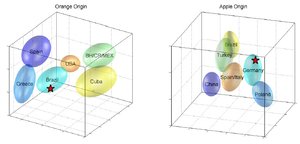
Rys. 4. Graficzne przedstawienie statystycznych modeli klasyfikacji dla określenia pochodzenia soku owocowego. Lewy rysunek przedstawia model dla soków pomarańczowych, prawy dla soków jabłkowych. Elipsoidy pokazują obszary dla grup referencyjnych, gwiazdka oznaczono badana próbkę [19].
Przytoczone przykłady i stosowane modele porównują różne grupy i ustalają podobieństwa w celu klasyfikacji próbki. Po określeniu najbardziej prawdopodobnej grupy przydziału, próbka jest dalej weryfikowana w dwóch krokach. Po pierwsze, jednowymiarowa analiza porównuje każdy przedział widma w stosunku do widma standardowego i określa odstępstwa od koncentracji związków. Rys. 5 (lewy widmo) pokazuje widmo poszerzone w obszarze około 2 ppm rozcieńczonego soku jabłkowego pochodzącego z Chin pokrywające się z populacją widmami referencyjnych (przedstawioną jako kolorowy rozkład w tle zmierzonego widma). Umożliwia to wykrywanie niezwykłych koncentracji składników (Rys. 5 widmo na prawym rysunku). Drugie podejście to wielowymiarowe analizy oparte na podejściu PCA/SIMCA (Soft Independent Modelling of Class Analogy) [7] do wykrywania odchyleń w analizie jednowymiarowej. Jeśli obie metody dają takie same rezultaty, próbka zgadza się z modelem, wtedy uważa się, że pomyślnie przeszła próbę badania wstępnego. W takim przypadku nie ma powodu badać dalej tej próbki. Jednakże jeśli wystąpiły odstępstwa potrzebne są dodatkowe badania z wykorzystaniem konwencjonalnych metod.
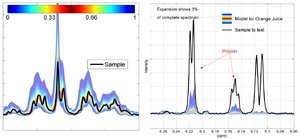
Rys. 5. Lewy rysunek: Weryfikacja próbki (czarna linia) w stosunku do grupy próbek referencyjnych (kolorowe tło) dla soku jabłkowego (~ 1 % widma); Prawy rysunek: sok pomarańczowy z zaznaczeniem wysokiej koncentracji związku wskazującego na intensywną ekstrakcję całych owoców łącznie z skórką [19].
Inną ważną metodą weryfikacji, szczególnie dla produktów występujących na rynku, jest oszacowanie zawartości owoców w soku. Konwencjonalnie jest to robione po przez ilościowe oszacowanie wyselekcjonowanych związków i minerałów i porównywane z referencyjnymi rozkładami.
Klasyfikacja SGF jest bardzo wydajną metodą analityczną do badania jakości soków owocowych. Szczególne parametry jakościowe soków owocowych mogą być zidentyfikowane dzięki zastosowaniu wielu testów statystycznych na jednym widmie. Metoda umożliwia rozróżnienie rodzajów owoców wykorzystanych do produkcji soku, dodawania cukru, wyróżnienia kraju lub regionu pochodzenia. Ponadto substancje obecne w soku mogą być określone ilościowo. Rutynowe zastosowanie metody klasyfikacji SGF daje także duże możliwości w częstości wykrywania odstępstw jakościowych. Efektywność metody umożliwia obniżenie kosztów analizy. Korzystając z osiągnięć metody klasyfikacji SGF można łatwo cała metodykę przenieść na inne zastosowania w badaniu jakości, przykładowo dla klasyfikacji mleka, wina lub piwa i innych rozpuszczalnych materiałów żywnościowych [20].
Autor: Magdalena Witek
1. http://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/Spectrpy/nmr/nmr1.htm. [cited.
2. Hrynkiewicz A. Z. and Rokita E., Magnetyczny rezonans jądrowy, in Fizyczne metody badań w biologii, medycynie i ochronie środowiska. 1999, Wydawnictwo Naukowe PWN SA: Warszawa. p. 115-135.
3. Nicholson J. K., Lindon J. C. and H. E., 'Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica, 1999. 29(11): p. 1181-1189.
4. Fiehn O., Kopka J., Dormann P., Altmann T., et al., Metabolite profiling for plant functional genomics. Nature Biotechnology, 2000. 18(11): p. 1157-1161.
5. Tang H. R. and W.Y. L., Metabonomics: a revolution in progress. Progress in Biochemistry and Biophysics, 2006. 33(5): p. 401-417.
6. Tang H. R. and W. Y. Nutrimetabonomics: metabonomics in food science. in The 8th Interantional Conference on the Application of Magnetic Resonance in Food Science. 2006. Nottingham: The Royal Society of Chemistry.
7. Kent J.T., Bibby J.M. and M. K.V., Multivariate Analysis. 1980, Maryland Heights, MO, USA: Academic Press Inc.
8. Ventura, D., Manifold Learning Examples PCA, LLE and ISOMAP. Tutorial PCA, 2008.
9. Spraul M., Freund A. S., Nast R. E., Withers R. S., et al., Advancing NMR sensitivity for LC-NMR-MS using a cryoflow probe: Application to the analysis of acetaminophen metabolites in urine. Analytical Chemistry, 2003. 75(6): p. 1536-1541.
10. Belton P. S., Delgadillo I., Holmes E., Nicholls A., et al., Use of high-field H-1 NMR spectroscopy for the analysis of liquid foods. Journal of Agricultural and Food Chemistry, 1996. 44(6): p. 1483-1487.
11. Belton P. S., Colquhoun I. J., Kemsley E. K., Delgadillo I., et al., Application of chemometrics to the H-1 NMR spectra of apple juices: discrimination between apple varieties. Food Chemistry, 1998. 61(1-2): p. 207-213.
12. Belton, P.S., I. Delgadillo, A.M. Gil, P. Roma, et al., High-field proton NMR studies of apple slices. Magnetic Resonance in Chemistry, 1997. 35: p. S52-S60.
13. Le Gall G., Puaud M. and C.I. J., Discrimination between orange juice and pulp wash by H-1 nuclear magnetic resonance spectroscopy: Identification of marker compounds. Journal of Agricultural and Food Chemistry, 2001. 49(2): p. 580-588.
14. Le Gall G., Colquhoun I. J. and D. M., Metabolite profiling using H-1 NMR spectroscopy for quality assessment of green tea, Camellia sinensis (L.). Journal of Agricultural and Food Chemistry, 2004. 52(4): p. 692-700.
15. Wang, Y.L., H.R. Tang, J.K. Nicholson, P.J. Hylands, et al., Metabolomic strategy for the classification and quality control of phytomedicine: A case study of chamomile flower (Matricaria recutita L.). Planta Medica, 2004. 70(3): p. 250-255.
16. Duarte I., Barros A., Belton P. S., Righelato R., et al., High-resolution nuclear magnetic resonance spectroscopy and multivariate analysis for the characterization of beer. Journal of Agricultural and Food Chemistry, 2002. 50(9): p. 2475-2481.
17. Rinke P., Moitrier S., Humpfer E., Keller S., et al., An 1H-NMR-Technique for high throughput screening in quality and authenticity control of fruit juice and fruit raw materials. Fruit Processing, 2007. 17: p. 10-18.
18. Spraul M., Humpfer E., Schäfer H., Schütz B., et al., NMR-Based Mixture Analysis on the Example of Fruit Juice Quality Control Using Statistics and Quantification., in NMR Spectroscopy in Pharmaceutical Analysis, U. Holzgrabe, I. Wawer, and B. Diehl, Editors. 2008, Elsevier: Amsterdam, The Netherlands. p. 319-339.
19. Spraul M., Schütz B., Rinke P., Koswig S., et al., NMR-Based Multi Parametric Quality Control of Fruit Juices: SGF Profiling. Nutrients, 2009. 1: p. 148-155.
20. Lachenmeier D., Humpfer E., Fang F., Schütz B., et al., NMR Spectroscopy for nontargeted screening and simultaneous quantification of health-relevant compounds in foods: The example of melamine. J. Agric. Food Chem., 2009. 57: p. 7194-7199.
Tagi:
magnetyczny rezonans jądrowy,
spektroskopia NMR,
metabonomika,
metabolomika,
SGF Profiling,
lab,
laboratorium,
laboratoria
wstecz
Podziel się ze znajomymi








Recenzje