
|

Zamknij X
|



Wykrywanie uszkodzeń w kwasie deoksyrybonukleinowym (DNA) na poziomie jednej (indywidualnej) komórki ma bardzo istotne znaczenie w takich dziedzinach jak: toksykologia, farmacja czy w diagnozowaniu chorób genetycznych. Ponadto, analize tą wykorzystuje się w testach genotoksyczności, a także w bio-monitoringu środowiska. Test kometkowy (SCGE-elektroforeza kometkowa) jest wszechstronnym, wrażliwym i prostym testem, stosowanym do pomiaru uszkodzeń i naprawy DNA w poszczególnych komórkach. Test ten jest bardzo wszechstronny, gdyż pozwala na ocenę uszkodzeń w różnych typach komórek i próbek tj. krwi obwodowej, komórek hodowanych, komórek nowotworowych, guzów litych, nasieniu, drożdżach czy bakteriach. Najpowszechniej stosowaną metodą jest test kometkowy przeprowadzany w warunkach alkalicznych [1].
Komórki osadzone na płytkach agarozowych poddaje się działaniu hipertonicznego roztworu do lizy i niejonowego detergentu, które to usuwają błony komórkowe, nukleoplazmę, cytoplazmę oraz powodują rozpuszczenie nukleosomów. Nastęnie, badane komórki traktowane są silnie zasadowym roztworem, powodującym relaksację („odprężenie”) cząsteczki DNA, dzięki czemu eksponowane są alkaliczne nietrwałe strony cząsteczki DNA (apurynowe i apirymidowe), które pojawiają sie jako przerwy w cząsteczce. Przerwy te w trakcie rozdziału elektroforetycznego migrują do anody pod wpływem przyłożonego prądu, co uwidacznia się w postaci tzw. „komet” [1].
Test kometkowy wykrywa uszkodzenia DNA w komórkach osadzonych w żelu agarozowym, poddanych lizie i denaturacji DNA, a następnie rozdziałowi elektroforetycznemu. Tak przygotowane komórki są barwione barwnikiem fluorescencyjnym wiążącym DNA, a następni e poddawane rejestracji obrazu w mikroskopie. W komórkach bez uszkodzenia DNA, DNA o wysokim ciężarze cząsteczkowym pozostaje związane z jądrem komórkowym. Obraz takiej komórki ujawnia się jako plamka na obrazach mikroskopowych. W komórkach, w których wystąpiły uszkodzenia DNA, powodujące pojawienie się przerw w niciach DNA, liza i denaturacja DNA prowadzi do odwijania superzwinietej struktury DNA, co z kolei pozwala na migrację DNA z jądra poddanego działaniu pola elektroforetycznego. Następnie, barwienie i analiza mikroskopowa ujawnia migrujące DNA jako ogon wydobywający się z jądra, dając wygląd bardzo podobny do astronomicznej komety, po którym test jest nazwany [2].
Przedstawiona poniżej procedura testu kometkowego podzielona zostałą na dwa dni: pierwszego dnia przygotowywane są próbki (komórki) do badania, płytki agarozowe, liza komórek, elektroforeza i neutralizacja, z kolei drugiego dnia przeprowadza się utrwalanie próbek , barwienie i analizę mikroskopową.
Dzień pierwszy: procedura pobierani a próbki krwi :
Około 1-2 ml ludzkiej krwi zbierane jest na heparynę jako antykoagulant w sterylnych warunkach, po czym natychmiast przeprowadza się separację limfocytów.
Oddzielanie limfocytów:
Do czystych i suchych probówek wirówkowych o pojemności 15 ml przenieść: 2 ml medium do oddzielania/wirowania limfocytów i około 1-2 ml pełnej krwi – nakładane w sposób uniemożliwiający mieszanie się obydwu skłądników. Tak przygotowane próbki poddawane są następnie wirowaniu ( 1500 obrotów/ minutę przez 30 minut, wirowanie prowadzone w temperaturze pokojowej). Pod koniec 30-minutowego wirowania pojawia się kożuszek zawierający jednojądrzaste komórki krwi obwodowej znajdujące się na granicy faz tj. pomiędzy plazmą i medium do oddzielania limfocytów, który jest aspirowany przy użyciu pipety pasteura do 1,5 ml probówki mikro-wirówkowej [1].
Należy mieć na uwadze, że test kometkowy przeprowadzany na świeżo izolowanych limfocytach daje lepsze wyniki. W celu optymalizacji wyników należy wykonywać minimum trzy powtórzenia na każdej z próbek, a dodatkowo powinny być stosowane kontrole wewnętrzne (pozytywne i negatywne) podczas wykonywania testu [1].
Przygotowanie preparatów: Żel agarozowy nakłada się na czyste i odtłuszczone szkiełka.
Przygotowanie agarozy:
0,5% agarozę o niskiej temperaturze topnienia oraz 0,75% agarozę o normalnej temperaturze topnienia miesza sie z buforem PBS i stopiono podgrzewa w mikrofalówce w ciągu 1-2 minut.
Zalecane jest, aby powyższe czynności wykonywać w ciągu dnia w słabym świetle, aby uniknąć uszkodzenia DNA wywołane promieniowaniem UV [1].
Pokrywanie szkiełek agarozą:
Należy pobrać ok. 100 ul gorącej 0,75% agarozy o normalnej temperaturze topnienia (NMPA), po czym nakropić ją na jednym końcu szkiełka. Kroplę rozetrzeć po całym szkiełku (wykorzystując w tym celu drugie szkiełko nachylone pod kątem około 45 °). Tak przygotowany „rozmaz” agarozy pozostawić do zastygnięcia w temperaturze 37 ° C. Przygotowanie pierwszej warstwy agarozy w powyższy sposób zapewnia lepsze zakotwiczenie dla kolejnych warstw agarozowym.
Nawarstwianie na szkiełko mieszaniny limfocyty - agaroza o niskiej temperaturze topnienia (LMPA)
Na zestaloną warstwę agarozy NMPA za pomoca pipety pasteura należy nanieść wcześniej przygotowaną mieszaninę: 60µL agarozy LMPA (37°C) z 20µL limfocytów (całość dokładnie wymieszana). Warstwę tą przykryć za pomocą szkiełka nakrywkowego, dzięki czemu tworzy się jednorodną warstwę na warstwie agarozy NMPA (czynnośc wykonywać powoli, by uniknąć powstania pęcherzyków powietrza). Przygotowane szkiełko pozostawia się do zestalenia w 4°C w lodówce (przez około 10-15 minut) [1].
Gdy warstwa limfocytów-LMPA zakrzepnie/zestali się,należy ostrożnie zdjąć szkiełko nakrywkowe, unikając oderwania z podstawowej warstwy. Następnie, dodać 75 ul agarozy LMPA na warstwę mieszaniny żelowej i przykryć świeżym szkiełkiem nakrywkowym ( unikając powstania pęcherzyków powietrza). Pozostawić do zestalenia żelu w 4 ° C w lodówce przez 10-15 minut [1].
W celu przygotowania roztworu nalezy zmieszać:
Gdy trzecia warstwa agarozy zestali się, delikatnie usuwa się szkiełko nakrywkowe, a płytki delikatnie zanurza się do barwienia zawierającego zimny bufor do lizy. Szkiełka przechowuje się w lodówce przez co najmniej 1 godz. Liza komórek może rozciągnąć się na więcej niż 24 godziny [1].
Roztwór I: rozpuścić 200 g wodorotlenku sodu (10 M) w 500 ml wody podwójnie destylowanej.
Roztwór II: możliwe jest również przygotowanie buforu w następujący sposób: rozpuścić 14,89 g soli disodowej EDTA (200mM) w 200 ml podwójnie destylowanej wody, po czym roztwór doprowadza się do pH=10 za pomocą wodorotlenku sodu [1].
Procedura alkalicznego odwijania i elektroforeza szkiełek
Po lizie w temperaturze 4 ° C, szkiełka delikatnie usuwa się z roztworu do lizy i ustawia dokładnie prostopadle do obu elektrod. Zbiornik do elektroforezy należy napełnić świeżym, zimnym buforem do elektroforezy - do momentu całkowitego zakrycia powierzchni preparatów (unikać tworzenia się pęcherzyków powietrza na żelu agarozowym). Szkiełka mogą być zanurzone w buforze zasadowym przez 30 minut w celu zrelaksowania nici DNA. Po czasie relaksacji rozpoczyna się rozdział elektroforetyczny, prowadzony przy zasilaniu równym 0,74 V / cm (między elektrodami) i 300mA. Rozdział elektroforetyczny prowadzony jest zazwyczaj przez 30 min.
Do rozdziału elektroforetycznego zawsze należy używać zimnego buforu do elektroforezy bądź prowadzić rozdział w warunkach chłodniczych. Działanie takie ma na celu uniknięcie uszkodzeń DNA powstałych ze względu na ciepło generowane podczas przepływu prądu [1].
W celu przygotowani buforu należy rozpuścić 48,5 g tris (0,4 M) w 800 ml podwójnie destylowanej wody. Wyregulować pH do 7,5 za pomocą stężonego kwasu solnego i dostosować do końcowej objętości 1000 ml.
Po rozdziale , szkiełka należy delikatnie podnieść z buforu elektroforetycznego. Przenieśc je na tacę do barwienia. Następnie, preparaty dokładnie zalać buforem do neutralizacji (bufor Tris o pH= 7,4). Inkubować je przez 5 minut,po czym usunąc bufor, a całą procedurę powtórzyć 2-krotnie. Na koniec szkiełka przemyć za pomocą wody destylowanej.
Barwienie szkiełek :
Kometki moga być uwidocznione na szkiełkach z wykorzystaniem barwienia barwnikami fluorescencyjnymi lub barwienia srebrem.
Fluorescencyjna metoda barwienia
Na każde z przygotowanych szkiełek nanosi się 50 ul barwnika bromku etydyny, po czym przykrywa się czystym szkiełkiem nakrywkowym. Do wizualizacji stosuje się mikroskop fluorescencyjny wyposażony w filtr wzbudzenia 515-560 nm z filtrem bariery 590 nm i powiększenie 200x. Co ważne, szkiełka barwione bromkiem etydyny nie mogą być przechowywane, a więc powinny być analizowane natychmiast po wybarwieniu [1].
Barwienie srebrem:
Po elektroforezie i neutralizacji, szkiełka suszy się przez noc. Następnie, umieszcza się je w roztworze utrwalającym na 10 minut, po czym po upływie czasu inkubacji szkiełka przemywa się kilka razy wodą destylowaną. Tak przygotowane pozostawia się do wyschnięcia w temperaturze 37 ° C przez co najmniej 1 godzinę (do maksimum ) przez noc przed barwieniem [1].
Odczynniki do barwienia srebrem:
Należy rozpuścić 75 g kwasu trichlorooctowego cynku, 25 g siarczanu cynku cynku i 25 g gliceryny w 400 ml podwójnie destylowanej wody. Całośc dokłądnie wymieszać przez 20-30 min. Po czym dopełnić woda do objętości 500 ml.
25 g węglanu sodu rozpuszcza się w 500 ml podwójnie destylowanej wody, przy ciągłym mieszaniu przez 20-30 min.
100 mg azotanu amonu rozpuszcza się z dodatkiem 100 mg azotanu srebra, 500 mg kwasu krzemowego i 250 µl formaldehydu w 500 ml podwójnie destylowanej wody. Zalecane jest aby roztwory barwiące A i B były przygotowywane zawsze na świeżo- bezpośrednio przed użyciem.
1ml lodowatego kwasu octowego należy zmieszać ze 100 ml wody podwójnie destylowanej [1].
Barwienie szkiełek:
Do barwienia szkiełek przygotowuje się mieszankę roztworów, w skłąd której wchodzi: 32 ml roztworu do barwienia A i 68 ml roztworu barwiącego B. Przygotowaną mieszankę wylewa się na szkiełka umieszczone wewnątrz pola barwienia (kolor bursztynowy) szkiełka barwione są w zamkniętym pudełku (pudełko z pokrywką) na kołysce, by zapewnić jednolite barwienie wszystkich szkiełek. Sam proces barwienia trwa ok. 10-20 min. ). Etap ten powtarza się 3-4 razy, za każdym razem używając świeżej mieszaniny roztworu A i roztworu B, a proces prowadzony jest do momentu pojawienia się szarawego zabarwienia na szkiełkach.Następnie, szkiełka przenosi się do roztworu zatrzymującego reakcję (inkubacja ok. 5 minut lub do momentu pojawienia się żółto-brązowego koloru.Na koniec, szkiełka przemywa się 1x wodą destylowaną i pozostawia do wyschnięcia w pozycji nachylonej, w temperaturze pokojowej [1].
Ocena uszkodzeń DNA
Uszkodzenie DNA można ocenić na kilka sposób tj. mierząc długość ogona komety lub optyczną ocenę stopnia uszkodzeń w skali od 0 do 4 w zależności od wyglądu komety. Alternatywnie, istnieją liczne programy do ilościowej analizy obrazu, które uwidaczniają dodatkowe parametry analizy, w tym m.in. procent uszkodzenia DNA, procent DNA w głowie komety, procent DNA w ogonie, moment ogona (tj. iloczyn długości ogona i procent DNA w ogonie). Na próbke analizowanych jest od 40 do 50 losowo wybranych komórek. Komety do analizy muszą być wybrane losowo z całej próbki (powinny reprezentować cały żel), zaś komety obserwowane na krawędziach preparatu (szkiełka) oraz preparaty zawierające pęcherzyki powietrza powstałe przez niewłaściwe nakładanie preparatu- powinny być odrzucone [1].
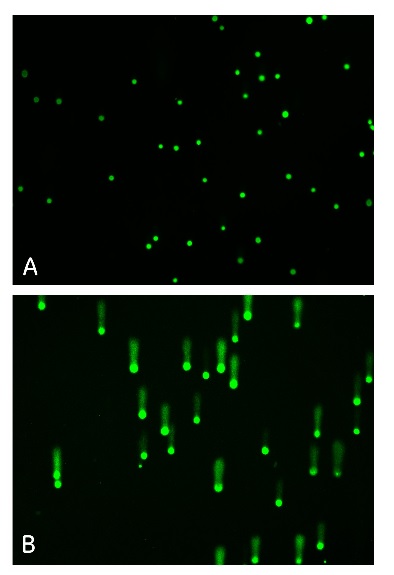
Zdjęcie: A- komórki nie traktowane nadtlenkiem wodoru, B- komórki inkubowane ze 100µM nadtlenkeim wodoru. A i B- wybarwione SybrGreen. Zdjęcie B- obraz kometek (Comet assay), https://promo.gelifesciences.com/na/k11286/misc/incell/Comet_Assay_SBS_Poster.pdf
Elektroforeza pojedynczych komórek w żelu (test kometkowy) jest jedną z najbardziej znanych metod stosowanych do pomiaru uszkodzeń DNA wywołanych stresem oksydacyjnym. Stosuje się ją m.in do oceny uszkodzeń w komórkach jednojądrzastych krwi obwodowej (PBMC) ,a także jako biomarker stresu oksydacyjnego w warunkach in vivo. Obróbka komórek do tesu tj. przechowywanie, usuwanie, obróbka i analiza próbek krwi wiąże się z ryzykiem tworzenia artefaktów, w związku z czym zazwyczaj próbki przygotowywane są bezpośrednio przed samą analizą (badaniem), bądź też poddaje się je wcześniej np. zamrożeniu (krioprezerwacja). Postępowanie takie jest czasochłonne, jednakże w swoich badaniach Al-Salmani K. i wsp. (2011) wykazali, że przechowywanie próbek krwi w małych objetościach (ok. 250 uL w temperaturze -80°C bez krioprezerwacji) przez okres 1 miesiąca nie wpływa na tworzenie się artefaktów uszkodzeń DNA. Przechowywanie dużych objętości krwi (np. 5 ml próbki) prowadzi do wzrostu uszkodzeń wraz z wydłużeniem czasu przechowywania- nawet w temperaturze -80°C i krioprezerwacji. Zastosowana metoda (Al-Salmani K. i wsp. (2011)) może mieć duże znaczenie dla pracy na próbkach archiwalnych, gdzie zazwyczaj dysponuje się małą ilością próbki [3].
Oksydacyjne uszkodzenia DNA są jednym z najbardziej rozpowszechnionych i mierzonych biomarkerów stresu oksydacyjnego. W trakcie wieloletnich badań nad DNA zauważono, że uszkodzenia DNA powstają również w trakcie ekstrakcji i obróbki DNA. W rezultacie, cała uwaga skupiła się na metodach analitycznych i warunkach przechowywania, które pozwoliłyby wyeliminować tego typu ryzyko. Pomimo, iż elektroofreza alkaliczna pojedynczych komórek ma też swoje wady, stała się jedną z popularniejszych metod stosowanych do oceny uszkodzeń powstających w DNA [3]. Sam problem zapobiegania generowania uszkodzeń w DNA w trakcie jego przechowywania jest nadal nie do końca rozwiązany [3].
Metoda comet assay pozwala na analizowanie rożnych tkanek, z których możliwe jest otrzymanie zawiesiny komórek . W zależności od tego jaki jest cel badania, wybrane komórki eksponowane są na testowane czynniki, bądź też analizie podlegają tzw. uszkodzenia endogenne. Uszkodzenia endogenne powstaja w trakcie normalnych lub patologicznych procesów metabolicznych- prowadząc do zmian w komórkach. Comet assay pozwala na:
Najprostsza procedura elektroforezy obejmu:
Przez lata badań wprowadzono różnorodne modyfikacje podstawowej metody comet assay, jednakże najbardziej znaną jest wersja tzw. elektroforezy alkalicznej , dzięki której możliwe jest mierzenie jednoniciowych pęknięc łańcucha DNA, a także wszelkich modyfikacji, które są niestabilne przy wysokim pH (głownie chodzi o miejsca AP). W tak zmienionej postaci comet assay wyparłą wcześniej stosowane metody analizy uszkodzeń DNA , w tym alkaliczną elucję, która wykorzystywana była przez wiele lat w badaniach toksykologicznych związanych z analizą pęknięć DNA [4].
Wykonanie analizy comet assay (wg procedury R. Słomski, Analiza DNA, 2008)
Roztwór lizujący:
Zmieszać kolejno ze sobą: 2,5 M NaCl (146,4 g/l), 0,1 M Na2EDTA (37,2 g/l), 10 mM Tris-HCl (1,2 g/l), pH=10, 1% Triton X-100 (10 ml/l- dodawany bezpośrednio przed lizą).
Roztwór elektroforetyczny:
300 m NaOH (12g/l), 1mM Na2EDTA (0,372 g/l).
Zdjęcie: odtłuszczanie szkiełka podstawowego,opalanie w płomieniu palnika http://www.uwm.edu.pl/wnz/v3/fck_files/file/mikrobiologia/MZ-TZrokII-cw2.pdf
Suche szkiełka zanurza się w gorącym roztworze 0,5% agarozie o normalnej topliwości ( w wodzie destylowanej), po czym jedną stronę szkiełek wyciera się bibułą ( w celu usunięcia agarozy), tak przygotowane szkiełka pozostawia się do wyschnięcia. Bardzo ważne na tym etapie jest zaznaczenie strony na której znajduje się agaroza, ponieważ po wyschnięciu strony szkiełek sa nie do rozróżnienia. Przygotowane w powyższy sposób szkiełka moga być bez ograniczeń przechowywane (w szczelnym pudełku). Do analizy comet assay zalecane jest stosowanie szkiełek podstawowych trawionych na całej powierzchni [4].
Pierwsza warstwa agarozy:
1% agaroza NMP w medium RPMI 1640 bez L-glutaminy (medium -podłoże RPMI 1640 stosowane jest jako podłoże wzrostowe i utrzymujące w hodowlach komórkowych, szczególnie stosowane jest do hodowli ludzkich limfocytów) [4], [6]. 80 µl roztworu agarozy nakrapia się na powierzchnię przygotowanej wcześniej suchej warstwy (jak wyżej), po czym przykrywa się szkiełkiem nakrywkowym. Po zestaleniu agarozy (po upływie ok. 5 minut, w temperaturze 4°C) szkiełko nakrywkowe należy delikatnie usunąc (tak by nie naruszyć warstw).
Druga warstwa agarozy:
1% agaroza o niskiej temperaturze topnienia (LMP) w podłożu RMPI 1640. 70 µl agarozy o temperaturze 37°C należy zmieszać z 30 µl zawiesiny komórek. Całość wylać na pierwszą (zastaloną) warstwę agarozy, po czym przykryć szkiełkiem nakrywkowym i schłodzić w 4°C. Po zestaleniu warstw (przed etapem lizy) należy ostrożnie zdjąć szkiełka nakrywkowe [4].
Liza: na tym etapie preparaty zalewa się schłodzonym (4°C) plynem lizującym w objętości okołu 15 µl na preparat, lizę prowadzić w temperaturze 4°C prze ok. 1 godzinę. Po upływie czasu inkubacji, roztwór lizujący jest usuwany a preparaty przenosi się do aparatu do elektroforezy.
Denaturacja alkaliczna: preparaty po lizie zalewa się schłodzonym (świeżo przygotowanym) roztworem elektroforetycznym, po czym preparaty poddaje się inkubacji prowadzonej w temperaturze nie przekraczającej 17°C. Po upływie 40 minut rozpoczyna sie rozdział elektroforetyczny. Warunki w jakich przeprowadzana jest elektroforeza zależą od odległości między elektrodami ). Optymalne stosowane napięcie mieści się w granicach 0,5 – 0,8 V/cm. Rozdział zazwyczaj prowadzi się w trakcie 30 minut przy 300 mA i napięciu 0,5V/cm. Należy mieć na uwadze fakt, że powtarzalność wyników w comet assay w dużej mierze zależy od przestrzegania przyjętych warunków elektroforezy.
Po rozdziale elektroforetycznym następuje etap neutralizacji, którą przeprowadza się z wykorzystaniem buforu neutralizującego. Rozdzielone żele neutralizuje się 3 razy po 5 min schłodzonym buforem. Następnie, przechodzi się do etapu barwienia. W tym celu na każdy żel nakrapia się 20 µl barwnika DAPI (o stężeniu 2µg/ml).
Analiza wyników: wybarwione żele analizuje się pod mikroskopem fluorescencyjnym w odpowiednim powiększeniu (200x z użyciem filtrów optycznych do DAPI). W zależności od posiadanego spzretu możliwe jest wizualne określenie liczby komet należących do czterech łatwo rozróżnialnych kategorii, mierzyć długośc komet lub ogonów, określać zawartość DNA w wybranych elementach komety, bądź też analizować bardziej złożone parametry (w tym np. tzw tail moment) [4]. Do pomiaru bardziej zaawansowanych parametrów ilościowych konieczne jest dysponowanie dodatkowym wyposażeniem w postaci kamery CCD. Kamera ta musi być sprzężona z komputerowym systemem cyfrowej analizy obrazu. Z kolei, określenie poszczególnych parametrów komety zależy od użytego opragramowania [7].
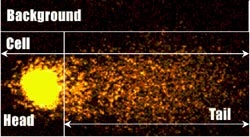
Zdjęcie: Typowa analiza budowy komety z uwzględnieniem głowy i ogona komety, a także tła, http://www.cometassayindia.org/definitions.htm
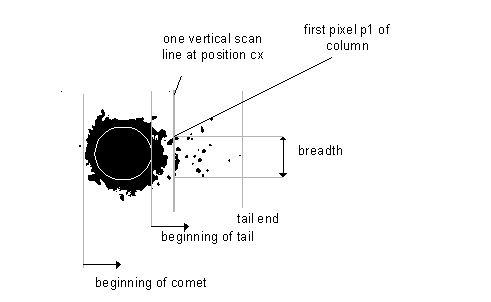
Do pomiarów prostych parametrów geometrycznych geometrycznych, w tym np. długości komety, wystarczy dowolny program umożliwiający tego rodzaju pomiary [4].
Elektroforeza alkaliczna pojedynczych komórek znana jest jako metoda szybka, prosta i czuła, dzięki czemu znalazła zastosowanie w wielu dziedzinach. Halder A. i wsp.(2002), wykorzystali comet assay do oceny uszkodzeń DNA spowodowanych chemioterapią stosowanej w ostrej białaczce limfoblastycznej. Pacjenci leczeni byli wg ustalonego planu. Do badań wykorzystano krew obwodową pacjentów pobraną na EDTA. Do analizy comet assay pobrano 1 ml heparynizowanej krwi. Test zakończono po 3 dniach od pobrania próbki. 20 µl krwi zmieszano z 1 ml lodowatego podłoża RMPI 1640 w probówce mikrowirókowej, a następnie izolowano limfocyty. Do izolacji limfocytów wykorzystano metodę z ficolem, podczas której otrzymuje się dwie fazy komórek, gdzie większość komórek nielimfoidalnych osiada na dnie probówki, natomiast limfocyty znajdują się na granicy gradientu (ficol) i osocza (interfaza). Tak otrzymane komórki znajdujące się na granicy gradientu i płynu nawarstwionego zbiera się bardzo ostrożnie i przepłukuje odpowiednim płynem odżywczym. Powyższa metoda izolacji opiera się na gradiencie gęstości, jej zaletą jest jednostopniowa procedura z równoczesnym uzyskiwaniem czystej frakcji limfocytów. Na otrzymanych komórkach wykonano test przeprowadzony według procedury Singh i wsp. (1988) z użyciem roztworu lizującego (2,5 M NaCL, 100 mM Na2EDTA, z dodatkiem świeżo przygotowanego 1% Triton X-100 oraz 10% DMSO), buforu elektroforetycznego (300 mM NaOH, 1mM Na2EDTA, pH=13 o temp. 4°C) oraz 0,6% agarozie LMP i NPM. Elektroforezę przeprowadzono przy 300 mA, a rozdzielone szkiełka barwiono bromkiem etydyny [8].
Analizę wyników wykonano wykorzystując mikroskop fluorescencyjny z 40x powiększeniem, wyposażonym w filtr wzbudzenia 515 – 560 nm oraz filtr ochronny 590 nm. Uszkodzenia DNA oceniano wzrokowo w sposób opisany przez IV (Palus i wsp., 1999), komórki sklasyfikowano w pięciu kategoriach : 0,I, II, III, IV (Palus i wsp., 1999). Z każdej próbki analizowano 100 komórek, które na postawie pomiaru średnicy sklasyfikowano jako:
Ze względu na fakt, że metoda comet assay stała się bardzo popularna, na rynku dostępnych jest coraz więcej kitów stosowanych do barwienia komórek po etapie elektroforezy. I tak, dostępne są zestawy do barwienia komórek srebrem (R&D system) bądź do barwienia fluorescencyjnego. Każde z barwień przeprowadzane jest w innych sposób, przy czym pierwszy typ (barwienie srebrem) wymaga analizy w mikroskopie optycznym, zaś drugi (barwienie fluorescencyjne)- użycia mikroskopu fluorescencyjnego. Obydwa sposoby barwienia dają porównywalne wyniki, sam obraz otrzymanych kometek jest także porównywalny (zdjęcie poniżej) [9].
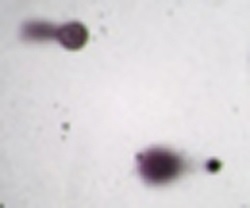
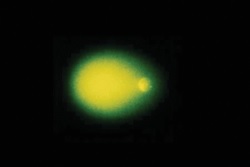
Zdjęcie: Barwienie kometek srebrem (lewa strona) oraz barwnikiem SYBR (prawa strona), http://www.rndsystems.com/product_detail_objectname_cometassay.aspx
Oporność na leki jest ogólnie uważana za główną przeszkodę udanej chemioterapii raka. Zjawisko to w protokołąch chemioterapii na ogół nie jest możliwe do przewidzenia (ani stopień zaawansowania oporności, ani czas się jej pojawienia). Z racji tego, że metoda comet assay bardzo się rozwija, ostatnie doniesienia mówią o możliwości zastosowania tej metody do identyfikacji i potencjalnego monitorowania reakcji komórek nowotworowych na różne leki stosowane w chemioterapii. Zasadniczo test ten (elektroforeza pojedynczej komórki) może być stosowany do dowolnego rodzaju guza, który leczony jest środkami chemioterapeutycznymi, które powodują jawne uszkodzenia w DNA.
Huang P. i wsp. (1998) w swoich badaniach testowali wpływ leków antynowotworowych (etopozyd) na uszkodzenia DNA, skupiając się na szerokim spektrum odporności na uszkodzenia DNA komórek nowotworowych. Oceniając komórki mysie (pochodzące z guzów od myszy z niedoborem odporności - rosnące jako monowarstwa) udało się, że określić że comet assay może być wykorzystana nie tylko jako wskaźnik wrażliwości na etopozyd, ale również można zademonstrować skuteczność (lub jej brak)w oporności wielolekowej (MDR - multidrug resistant) [10].
Huang P. i wsp. w swoich badaniach do oceny uszkodzeń DNA zastosowali alkaliczny test kometkowy [10].
Etopozyd jest lekiem cytostatycznym, który stosowanym jest w leczeniu nowotworu płuc, jajników oraz jąder. W trakcie chemioterapii lek ten możne być podawany kilkoma drogami:
- przez wlew dożylny, przez wenflon umieszczony w żyle, zazwyczaj na grzbiecie dłoni,
- wkłucie centralne (drobna, plastikowa rurka wprowadzona pod skórę do żyły w okolicy obojczyka)
- wkłucie obwodowe (drobna plastikowa rurka wprowadzona do żyły w pobliżu zgięcia łokciowego),
- doustnie, w kapsułkach. Wlew leku trwa zazwyczaj w granicach 30 - 60 minut.
Chemioterapia zazwyczaj podawana jest w postaci kilku cykli- przez okres kilku miesięcy. Czas trwania leczenia i liczba cykli zależy od rodzaju zdiagnozowanego nowotworu [11].
Procedura comet assay wg. Huang P. i wsp (1998)
Komórki zebrano w zawiesinę 2x104 komórek ml-1 i przetrzymywano w 4°C. 0,5 ml próbki przeniesiono do jednorazowych probówek, po czym dodano do nich 1,5 ml 1% agarozy o niskiej temperaturze topnienia. Następnie, z przygotowanego mieszaniny szybko odpipetowano 1,5 ml i naniesiono na półmatowe szkiełko mikroskopowe, po czym przeniesiono je w chłodne miejsce . Następnie, szkiełka zanurzono na 1 godzinę w alkalicznym buforze lizującym zawierającym 1M NaCl, 0,03M wodorotlenek sodu oraz 0,2% laurylo-sarkozynę (utrzymując szkiełka w położeniu poziomym).
Po upływie czasu inkubacji szkiełka przemyto 2x 0,03M wodorotlenkiem sodu z dodatkiem 2mM EDTA. Następnie, szkiełka poddano elektroforezie przy 0,6V cm-1 przez 25 minut w świeżym buforze do elektroforezy zawierającym: 0,03M wodorotlenek sodu oraz 2mM EDTA (rozdział poziomy). Po rozdziale szkiełka przepłukano wodą i poddano 20-minutowemu barwieniu z wykorzystaniem jodku propidyny (2,5µg ml-1). Na końcu analizie w mikroskopie fluorescencyjnym poddano ok. 200 indywidualnych komórek (kometek) z każdej przygotowanej próbki. Uszkodzenia DNA analizowo utomatycznie dzięki dołączonemu specjalnemu oprogramowaniu [11].
Połączenie comet assay z metodą FISH
FISH (Fluorescence In Situ Hybrydisation) to metoda polegająca na połączeniu (tzw. hybrydyzacji) sondy molekularnej z komplementarnym DNA w chromosomach lub jądrach interfazowych na preparatach mikroskopowych. Dzięki zastosowaniu znakowania sondy za pomocą fluorochromów, powstałe miejsca hybrydyzacji mogą być następnie analizowane pod mikroskopem i lokalizowane w chromosomach. Bardzo ważne w tej metodzie jest to, że można stosować kilka sond jednocześnie. Ogromny postęp jaki poczynił sie zarówno w mikroskopii fluorescencyjnej, jak rónież komputerowej analizie obrazu i immunocytochemii, umożliwia opracowywanie wielu różnych modyfikacji metody FISH, pozwalających na wykrywanie coraz krótszych fragmentów DNA i stosowaniu większej liczby sond równocześnie. Jedną ze znananych modyfikacji jest połączenie metody FISH z analizą kometek (comet assay) [13].
Połączenie tych dwóch jakże użytecznych i dobrze znanych metod, pozwala na wykrywanie uszkodzeń w DNA w szczególnych regionach genów w stosunku do całego genomu. Metoda ta została wykorzystana w wielu badaniach, gdzie z powodzeniem zlokalizowano uszkodzenia DNA wewnątrz komet -używając specyficznych sond (sondy specyficzne dla genu i chromosomu). W metodzie FISH wykorzystuje się sondy specyficzne dla chromosomu . Sondy te pozwalają na wykrycie zmian w chromosomie dotyczących bardzo małego jego odcinka. Tego typu sondy otrzymywane są na drodze laserowego wycinania poszczególnych fragmentów chromosomu i ich fluorescencyjnego znakowania [13], [14]. Połączenie FISH z comet assay jest bardzo przydatne, ponieważ zdolność uzyskania tego rodzaju informacji przyczynia się do tego, że test ten może być wykorzystany w wielu różnych dziedzinach nauki, w tym. m.in. w wielu obszarach badań klinicznych, dostarczając cennych informacji na temat swoistych cech DNA z pojedynczych komórek oraz ich reakcji na różne czynniki zewnętrzne (promieniowanie, chemikalia czy leki). Informacje te są szczególnie przydatne w diagnozowaniu, prognozowaniu i leczeniu raka, umożliwiając analizę komórek nowotworowych [14].
McKenna i wsp. (2012) pzreprowadzili badania, w których wykorzystali połączenie metody comet assay z FISH. W tym celu, jako pierwszy etap przeprowadzono alkaliczną elektroforezę według standardowego protokołu McKelvy-Martin i wsp. (2009). Komórki z hodowli przemyto dwukrotnie w 10 ml soli fizjologicznej buforowanej fosforanem (PBS ) . Żywotność komórek oceniano stosując metodę wykluczenia błękitem trypanowym . We wszystkich eksperymentach żywotność komórek wynosiła> 99%. Następnie, pobrano 1ml komórek zawieszonych w Ca2 + i Mg2 + (bez PBS), w stężeniu 2 x 10 5 komórek / ml za pomocą pipety, i przeniesiono do probówki typu Eppendorf. Próbkę zwirowano przy 1500 rpm przez 5 minut w temperaturze 4 ° C. Po wirowaniu, na szkiełka mikroskopowe naniesiono po 100µL 0,6% agarozy o normalnej temperaturze topnienia, przygotowanej w Ca2+ i Mg2+ (bez PBS) w 37°C . Powleczone agarozą szkiełko przykryto szkiełkiem nakrywkowym i przeniesiono na lód w celu zestalenia agarozy.Na kolejnym etapie przygotowano agarozę o niskiej temperaturze topnienia (1,2%), którą następnie zmieszano w stosunku 1:1 z pożywką hodowlaną (zawierającą 20% FBS), po czym 80uL otrzymanej mieszaniny użyto do rozpuszczenia otrzymanej po wirowaniu peletki komórek. Ze szkiełka z agarozą delikatnie usunięto szkiełko nakrywkowe i od razu (na pierwszą warstwę agarozy) naniesiona pipetą mieszaninę agarozy z komórkami . Szkiełko ponownie nakryto świeżym szkiełkiem nakrywkowym, a całość pozostawiono do zestalenia na lodzie [14]. Po zdjęciu szkiełka nakrywkowego, preparat napromieniowano, po czym umieszczono w roztworze do lizy (2,5 M NaCl , 100 mM Na2EDTA , 10 mM Tris , pH 10 , 1% Triton X -100 dodano) na 1 godzinę w temperaturze 4 ° C. Po lizie szkiełka poddano elektroforezie poziomej - zbiornik do elektroforezy napełniono świeżym, schłodzonym buforem do elektroforezy (300 mM NaOH, 1 mM Na2EDTA, pH> 13). Szkiełka pozostawiono na 20 minut w buforze w celu umożliwienia rozwijania DNA, po czym rozpoczęto rozdział elektroforetyczny (25 V (0,66 V / cm) i 300 mA przez 20 minut). Po rozdziale preparaty neutralizowano 3 razy, płukając je po 5 minut w 2X SSC (3 M roztwór soli cytrynianu sodu; 0,3 M cytrynian sodu, pH 5,3). Po płukaniu, szkiełka osuszono i odwodniono przez 2-minutowe płukanie w roztworach o wzrastającym stężeniu etanolu (70%, 85%, 100%) . Na koniec szkiełka suszono na powietrzu [14].
Przygotowanie sond i hybrydyzacja (comet assay-FISH)
Na preparatach z kometkami przygotowano FISH, używając mieszaniny dwóch znakowanych sond, zmieszanych w równych stężeniach. Na każde szkiełko z kometami naniesiono mieszaninę hybrydyzacyjną zawierając równe stężenia sond, po czym preparaty nakryto szkiełkiem nakrywkowym. Szkiełka zdenaturowano w 80°C przez 2 minuty. Hybrydyzację przeprowadzano w temperaturze 37°C przez 16 godzin, w ciemnej i wilgotnej komorze hybrydyzacyjnej [14].
Post-hybrydyzacja i kontrastowanie
Po hybrydyzacji, szkiełka umieszczono w 50% roztworze formamidu i 2X SSC (10 minut w temperaturze 45 ° C)- szkiełka delikatnie mieszano w celu odczepiania szkiełek nakrywkowych. Przemywanie powtórzono 3 razy, a na koniec szkiełka inkubowano 10 minut w 2X SSC w temperaturze 45 ° C, oraz 5 minut w 2X SSC zawierającym 0,1% Igepal (Sigma). Szkiełka pozostawiono do wyschnięcia na powietrzu przez 30 minut, po czym podbarwiono 16 ul roztworu DAPI [14]. Otrzymane w powyższy sposób szkiełka pozostawiono w ciemności w temperaturze 4°C przez czas nie dłuższy niż 2 godziny przed obserwacją. Cała procedura wykonywana była w żółtym świetle, by uniknąć dalszych uszkodzeń DNA w naturalnym świetle [14].
Comet assay-FISH (analiza)
Obserwacje szkiełek przeprowadzono w końcowym powiększeniu x600, z wykorzystaniem mikroskopu epifluorescencyjnego podłączonego do kamer. Wykorzystano potrójny filtr pasmowy: DAPI (wzbudzenie 370 nm, emisja 450 nm, szerokość pasma 20 nm), widmo pomarańczowe (wzbudzenie 560 nm, emisja 590 nm, szerokość pasma 60 nm) i widmo zielone (wzbudzenie 547 nm, emisji 572 nm, szerokość pasma 30 nm) [14].
Autor: Lidia Koperwas
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje