
|

Zamknij X
|




Fosfatazy są enzymami należacymi do klasy hydrolaz, których funkacja polega na hydrolizowaniu estrów kwasu fosforowego, a dzięki temu na uwalnianiu fosforu nieorganicznego (Pi) ze związków organicznych. Enzymy te są szeroko rozpowszechnione w organizmie, w wysokich stężeniach obecne są w kościach, jelitach, nerkach, wątrobie i łożysku. Stężenie enzymu (normalnie występującego w osoczu) uzależnione jest przede wszystkim od watroby i jelit oraz w niewielkim stopniu od kości. Wzrost aktywności fosfatazy zasadowej w surowicy jest ważnym wskaźnikiem zaburzeń wątroby i dróg żółciowych lub kości [1], [5].
Fosfataza alkaliczna ALP jest enzymem szeroko rozpowszechnionym w organizmie ludzkim: u dorosłych częściowo pochodzi z wątroby (frakcja termostabilna) i częściowo z kości, RES i układu naczyniowego (frakcja termolabilna). Dzięki temu też wyróżniane są jej różne izoenzymy. Aktywność osoczowa frakcji kostnej fosfatazy alkalicznej w normalnych warunkach osiąga maksymalne wartości u dzieci w trakcie ich wzrostu (do trzykrotnej wartości poziomu organizmu dorosłego), w związku z tym izoenzym ten występuje również w osteoblastach (komórkach związanych z procesem formowania kości i wbudowywania wapnia). Pod koniec pierwszego trymestru ciąży zauważa się wzrost produkcji enzymu, co jest reakcją fizjologiczną związaną ze wzrostem izoenzymu łożyskowego, który osiąga najwyższy poziom w tym okresie (ok. dwukrotnie ponad normę).
Niestety, oprócz wartości fizjologicznych, rozróżnia się także patologie związane z podniesieniem się aktywności surowiczej fosfatazy alkalicznej, wśród których diagnozuje się m.in.: przerzuty nowotworowe do kości i wątroby (produkujące enzymy), żółtaczkę zewnątrz i wewnątrzwątrobową, zespoły upośledzonego wchłaniania z nadżerkami i wrzodami błony śluzowej czy uszkodzenia w procesach zdrowienia takich jak: ostry zawał mięśnia sercowego, płuc czy nerek [13].
Wszystkie fosfatazy podzielono na 3 głowne grupy, wyszczególnione ze względu na ilość wiązań estrowych danego substratu:
- fosfomonoesterazy (określane również jako hydrolazy monoestrów fosforanowych)
- fosfodiesterazy (hydrolazy diestrów fosforanowych)
- fosfotriesterazy (hydrolazy triestrów fosforanowych) [1].
Ponadto, w zależności od pH działania, enzymy podzielono na kwaśne fosfomonoesterazy (kwaśne fosfatazy) oraz alkaliczne fosfomonoesterazy (alkaliczne fosfatazy). Fosfatazy kwaśne wykazują maksimum działąnia przy pH=4 – 6, zaś zasadowe mają optimum pH w granicach 8-10 [1]. Ze względu na lokalizację wyróżnia się fosfatazy wewnątrzkomórkowe oraz zewnątrzkomórkowe (tzw. fosfatazy sekrecyjne). Fosfatazy wewnątrzkomórkowe uczestniczą w remobilizacji Pi ze źródeł wewnętrznych. Występują one zarówno w wegetatywnych, jak i generatywnych organach roślin (głównie w wakuolach). Fosfatazy sekrecyjne uczestniczą w pozyskiwaniu fosforu z podłoża [1].
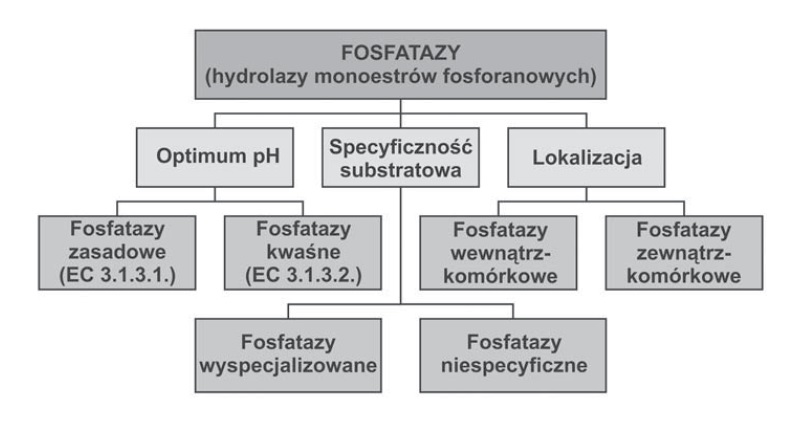
Zdjęcie: Podział fosfataz [1].
Oznaczanie aktywności fosfataz
Do określania aktywności tych enzymów wykorzystuje się zazwyczaj syntetycznie otrzymane substraty, a wśród nich najczęściej są to: α i β-glicerofosforan, fenylofosforan, ρ-nitrofenylofosforan czy ester fosforowy fenoloftaleiny. Aktywność mierzy się na podstawie obserwacji dwóch typów, tj.:
1)ilością uwolnionego nieorganicznego fosforu lub
2)ilością uwolnionej reszty organicznej estru fosforanowego po inkubacji enzymu z substratem, a cała reakcja przeprowadzana jest w ściśle określonych warunkach. Zastosowanie tej metody uzależnione jest od budowy chemicznej związku. Fenol oznaczany jest z wykorzystaniem odczynnika Folina-Ciocalteu, z kolei fenoloftaleinę i p-nitrofenol – po zalkalizowaniu próbki, zaś np. naftol oznaczany jest kolorymetrycznie po sprzęgnięciu z solą diazoniową na barwnik diazowy [3].
Sole diazoniowe powstają w reakcji amin aromatycznych z kwasem azotowym. Stałe sole diazoniowe są bardzo niebezpieczne, wybuchowe, a niektóre z nich są również kancerogenne. Reakcja diazowania przebiega w środowisku kwaśnym, tak więc amina, która bierze udział w takiej reakcji ulega rozpuszczeniu w roztworze kwasu solnego lub kwasu siarkowego, a następnie tworzy chlorowodorek lub siarczan. Reakcja taka przebiega według wzoru: C6H5N2+ + H2O → C6H5OH + N2(g) + H+ [2], [3].
Otrzymywanie soli diazoniowych
Sole diazoniowe powstają w reakcji fenyloaminy (zznanej rónież jako anilina lub aminobenzen) z kwasem azotowym – zwłaszcza w reakcji w temperaturze poniżej 5°C.

Kwas azotowy ulega bardzo szybko rozłożeniu. W przypadku reakcji z fenyloaminą, fenyloamina najpierw ulega rozpuszczeniu w kwasie solnym, a następnie dodaje się roztwór azotynu sodu lub azotyn potasu. W reakcji między kwasem solnym i jonami azotowymi powstaje kwas azotowy. Fenyloaminy reagują z kwasem azotowym w różny sposób, w zależności od temperatury reakcji [4].
Reakcja z ogrzewaniem mieszaniny
Jeżeli mieszaninę ogrzewa się, można uzyskać czarny oleisty produkt zawierający między innymi fenol i wydzielając się (w postaci gazu) azot:

Reakcja w niskich temperaturach
Wszystkie roztwory należy ochłodzić w zlewkach z lodem tj.: roztwór fenyloaminy z kwasem solnym (chlorek fenyloaminy) oraz roztwór azotynu sodu lub potasu (przetrzymywany w tych samych warunkach). Następnie, ochłodzony roztwór azotynu dodaje się do roztworu chlorku fenyloaminy, w taki sposób by temperatura roztworów nie wzrosła powyżej 5°C. W wyniku reakcji powstaje chlorek benzenodiazoniowy.
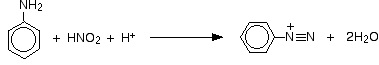
Jon dodatni zawierający grupę –N2+ znany jest jako jon diazoniowy. Część nazwy „azo” odnosi się do azotu [4].
Mechanizm działania fosfataz
Proces działania enzymów jest dobrze poznany, dzięki czemu wiadomo, że polega on na sekwencyjnym uwalnianiu produktów hydrolizy monoestrów fosforanowych. W trakcie reakcji pierwszy uwalniany jest produkt alkoholowy, który albo nie wykazuje hamowania aktywności enzymów, albo jest to hamowanie niekompetycyjne. Jako końcowy produkt reakcji uwalniany jest fosfor (Pi), który hamuje enzym kompetycyjnie. Aktywność kwaśnych fosfataz może być stymulowana dwuwartościowymi kationami metali, takimi jak: Ni2+, Ca2+, Mg2+. Hamowanie aktywności enzymów zachodzi w obecności jonów metali ciężkich czy związków takich jak: arsenian, molibdenian, winian bądź szczawian [1].
Zestaw do pomiaru aktywności fosfatazy kwasowej (King & King’s Method (King. P.R.M & King, E.J. Clin Path 7.322.), M/s Excel Diagnostics, procedura pochodząca ze strony: http://exceldiag.com/catalogs/10003.pdf)).
Do określania aktywności fosfatazy kwasowej można wykorzystać kilka różych substratów np.: p-nitrofenylofosforan. W metodzie King’a wykorzystuje się fenylofosforan disodowy, który jest hydrolizowany przez fosfatazę kwaśną z wydzieleniem fenolu, a metoda ta jest szeroko stosowana. Fenol uwalniany jest proporcjonalnie do aktywności fosfatazy, która mierzona jest kolorymetrycznie. Ponadto, fenol jest związkeim reaktywnym, dzięki czemu może być określany wrażliwymi metodami kolorymetrycznymi, umożliwiając w ten sposób skrócenie czasu inkubacji [7], [8].
Firma M/s Excel Diagnostics Pvt.Ltd wprowadziła zestaw do pomiaru aktywności kwaśnej fosfatazy oparty na zmodyfikowanej metodzie Kinga (King & King’s Method), w której buforowany substrat jest specjalnie stabilizowany i przedstawiony jest jako fiolkowy monotest. Ponadto, kolor reagent jest zmodyfikowany, w celu zmniejszenia dodawanej ilości, tak więc dodatki odczynników minimalizują błędy podczas wykonywania testu [7].
Zasada metody:
fosfataza kwaśna z surowicy ulega hydrolizie do fenolu i wodorofosforanu disodowego, przy pH=10. Tak wytworzony fenol poddaje się reakcji z 4-aminoantypiryną w środowisku zasadowym, w obecności środka utleniającego, którym jest żelazocyjanek potasu z utworzeniem czerwonego kompleksu, którego absorbancja jest wprost proporcjonalna do aktywności enzymu [7]. Oznaczenie próbek wykonuje się długości fali równej 510 nm (zielony filte), po wcześneijszej inkubacji próbek w 37°C w czasie od 3-15 minut [7].
Wykonanie oznaczenia z zastosowaniem zestawu M/s Excel Diagnostics (King & King’s Methods; King. P.R.M & King, E.J. Clin Path 7.322.)

Zdjęcie: http://exceldiag.com/catalogs/10003.pdf
ALP w jednostkach KA unit/dl= A(Test) – A(Control) / A(Standard) – A(Blank) x 10 (Std. Conc.)
1KA unit/dl = 7.1 U/l [7].Oznaczanie aktywności fosfatazy zasadowej i kwasowej
W doświadczeniu wykorzystuje się metodę Kinga-Armstronga (1965). Zasada metody opiera się na hydrolizie fenylofosforaniu disodowego przez fosfatazę, a ilość wytworzonego w reakcji fenolu jest miarą aktywności enzymu. Do oznaczenia fenolu wykorzystuje się odczynnik Folina-Ciocalteu w metodzie kolorymetrycznej. Otrzymany wynik podawany jest w jednostkach Kinga-Armstronga [3].
Jednostka Kinga-Armstronga oznacza mg fenolu uwolnionego z fenylofosforanu disodowego przez enzym w 100 ml surowicy , w temperaturze 37°C, w ciągu 15 minut przy pH=10 dla fosfatazy zasadowej, oraz w ciągu 60 minut przy pH=4,9 dla fosfatazy kwasowej [3].
Wykonanie:
Do probówek wirówkowych należy odmierzyć po 2 ml buforu węglanowo-wodorowęglanowego o pH=10 dla fosfatazy zasadowej (tj.: rozpuścić 6,36 g Na2CO3 oraz 3,36 g NaHCO3 w wodzie i uzupełnić wodą do objętości równej 100 ml) lub buforu cytrynianowego o pH=4,9 dla fosfatazy kwasowej (tj.: rozpuścić 21 g kwasu cytrynowego w wodzie, dodać 188 ml 1M roztoru NaOH, uzupełnić wodą do obj. 500 ml. Sprawdzić pH- w razie potrzeby doprowadzić do odpowiedniego pH za pomocą 1M roztworu HCl lub NaOH. Na koniec do roztworu dodać kilka kropli chloroformu i przechowywać w lodówce) oraz 2 ml substratu. Probówki ogrzewać kilka minut w łaźni wodnej o temperaturze 37°C, po czym dodać po 0,2 ml surowicy. Dobrze wymieszać i inkubować w temp. 37°C przez 15 minut (oznaczenie aktywności fosfatazy zasadowej) lub przez 60 minut (oznaczenie aktywności fosfatazy kwasowej). Po inkubacji do próbek dodać 1,8 ml rozcieńczonego (1:2) odczynnika Folina-Ciocalteu, całość wymieszać , a po kilku minutach inkubacji próbki odwirować. Po wirowaniu z próbek pobrać po 4 ml supernatantu (do wywołania barwy) [3].
Jednocześnie wykonać próbę kontrolną: do 2 ml buforu oraz 2 ml substratu należy dodać 0,2 ml surowicy i od razu 1,8 ml rozcieńczonego odczynnika Folina-Ciocalteu, próbkę wymieszać i odwirować, po czym pobrać 4 ml supernatantu [3].
Do probówek z supernatantami dodać po 2 ml 15% Na2CO3, inkubować w łaźni wodnej o temperaturze 37°C przez 10 minut. Po inkubacji należy oznaczyć absorbancję dla próbek wobec wody przy długości fali równej λ=680 nm [3].
Przygotowanie krzywej kalibracyjnej:
Jako pierwszy etap należy przygotować szereg rozcieńczeń wzorcowych roztworu fenolu (tj.: macierzysty roztwór wzorcowy fenolu zawierający 1mg fenolu w 1 ml (0,1% fenolu w 0,1M HCl)). Rozcieńczenia przygotowuje się tak, by uzyskać stężenia od 0,002 mg do 0,02 mg w 1 ml.
Do 2 ml przygotowanych rozcieńczeń dodać 1,2 ml odczynnika Folina-Ciocalteu, 0,8 ml wody oraz 2 ml 15% roztworu Na2CO3. Próbki wymieszać i inkubować w łaźni wodnej w temperaturze 37°C przez 10 minut. Po upływie czasu inkubacji, odczytać absorbancję wobec próby kontrolnej na odczynniki i wykreslić krzywą kalibracyjną dla mg fenolu [3].
Obliczenia do doświadczenia:
Wartość absorbancji próby kontrolnej odejmuje się od wartości próby badanej, a następnie z krzywej kalibracyjnej należy odczytać ilość uwolnionego fenolu (w mg). Otrzymany wynik mnoży się wartość 750 (do wywołania barwy w doświadczeniu pobrano po 4 ml z 6 ml inkubatu, który zawiera 0,2 ml surowicy, uwzględniając te dane, otrzymuje się powyższą artośc tj.: (100/0,2) x (6/4)=750). Wartość wyniku odczytuje się w jednostkach Kinga-Armstronga (K-A) [3].
Jednostka Kinga-Armstronga w układzie SI
Rozwój gałęzi medycyny laboratoryjnej jest ściśle związany z rozwojem metod badawczych, a także technik pomiarowych, które wykorzystywane są do wykonywania badań. Wraz z rozwojem wiedzy z dziedzin takich jak: chemia, fizyka oraz nauki przyrodnicze i medyczne, ewoluowały także jednostki, w których wyrażano wyniki pomiarów badań laboratoryjnych. I tak, przełomem była zamiana związana z wyrażaniem stężenia elektrolitów we krwi . Używana „od zawsze” jednostka miligramów na decylitr (mg/dl) została zastąpiona jednostkę miliekwiwalenty na litr (mEq/l). Zamiana ta dotyczyła oznaczania stężenia sodu, potasu oraz chlorków. Co więcej, w istotny sposób wpłynęła ona na sposób interpretacji wyników oznaczeń elektrolitów. Sam fakt wprowadzenia nowej jednostki był ściśle związan z możliwością oceny stanu gospodarki wodno-elektrolitowej ustroju, z jednoczesnym uwzględnieniem zasady tzw. elektroobojętności płynów ustrojowych. Aktualnie,większość laboratoriów medycznych dla wyrażania stężenia elektrolitów czyli np. sód czy potas, stosuje jednostkę mmol/l, która jest jednostką układu SI [6].
W toku „ewolucji jednostek”, zmieniono również sposób wyrażania aktywności katalitycznej białek enzymatycznych. Początkowo aktywność enzymu wyrażana była w bardzo różnych jednostkach, co w głównej mierze związane było z wykorzystywaniem różnych metod badawczych oznaczania aktywności katalitycznej enzymu. Najczęściej stosowana nazwa jednostki pochodziła od nazwiska badacza, który opracował daną metodę. I tak, aktywność katalityczną amylazy, fosfatazy kwaśnej i zasadowej wyrażano w jednostkach umownych. Aktywność amylazy wyrażana była w jednostkach bez nazwy, jednakże za każdym razem obok wyniku podawano nazwę metody, którą wykonano dane oznaczenie, np.:
Do wyrażania aktywności fosfatazy zasadowej i kwaśnej stosowano kilka róznych jednostek, m.in.:
1. jednostkę Bodansky’ego (od stosowanej metody Bodansky’ego)
2. jednostkę Kinga-Armstronga (metoda Kinga-Armstronga)
3. jednostkę Besseya (metoda Besseya) [6].
W niektórych przypadkach istniały wzajemne przeliczniki jednostek umownych, dzięki czemu dany wynik można było podać w dwóch jednostkach,np.: 1 jednostka Besseya odpowiadała ok. 1,8 jednostek Bodansky’ego. Stosowanie wielu jednostek umownych,było niestety nie do końca dobrym rozwiązaniem, gdyż uniemożliwiało to lub utrudniało porównywanie wyników badań, które zostały uzyskane w różnych jednostkach badawczych - laboratoriach. W konsekwencji ciągłego dążenia do doskonalenia metod oznaczania aktywności enzymów było przyjęcie ujednoliconej jednostki służącej do wyrażania aktywności enzymów. W 1961 roku Międzynarodowa Unia Biochemii i Biologii Molekularnej wprowadziła standardową jednostkę aktywności enzymów, którą nazwano jednostką międzynarodową (ang. international unit, IU). Przyjętą jednostkę zdefiniowano jako” „aktywność katalityczną zdolną do przekształcenia 1 mikromola substratu w czasie 1 minuty”. Ponadto, zalecono, by aktywność enzymów wyrażana była w jednostkach na litr (IU/l) lub w milijednostkach na mililitr (mIU/ml) [6].
E. J. King i wsp. w 1951 roku, w oparciu o pierwotną metodę King’a i Armstrong’a (1934), która wykorzystywała fenylofosforan disodowy hydrolizowany przez fosfatazę kwaśną z uwolnieniem fenolu, opracowali dwie podobne metody oznaczania aktywności fosfatazy. Głównym powodem opracowania poniższych metod, w których niezwykle ważny jest końcowy pomiaru ilości uwolnionego nieorganicznego fosforanu był fakt, że oszacowanie fosforanu w osoczu było często wymagane w badaniu chorób u dzieci. Uważano,że pożądane jest łączenie oszacowania ilości uwolnionego fosforanu z określeniem ilości fosfatazy, co może być łatwo wykonane poprzez pomiar nieorganicznego fosforanu z identyczną kontrolą dla fosfatazy. Pomiary takie przeprowadzane były z wykorzystaniem niektórych procedur np. Jenner i Kay (1932)i Bodansky (1933). Jednakże, zastosowanie fenylofosforanu do oznaczania fosfatazy było bardziej preferowane, z powodu większej wygody procedury- przebiegała w znacznie krótszym czasie, co było niezbędne do wykonania oznaczenia w momencie dysponowania małymi ilościami osocza lub surowicy [9].
Metoda I (wg ]. King E.J i wsp. (1951), http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1023361/?page=1)
Odczynniki wykorzystane w procedurze:
1)Substrat: W poniższej metodzie jako substrat wykorzystuje się 0,01 M fenylofosforan disodowy, otrzymany przez rozpuszczenie 2,18 g w 1 litrze wody destylowanej, która jest szybko ogrzewana do wrzenia (w celu zniszczenia drobnoustrojów). Roztwór należy przechowywać na lodzie.Dodatek kilku kropli chloroformu działa jak środek antyseptyczny.
2)Bufor (pH = 10): 0,1 M roztwór 6Na2CO3: 4NaHCO3 przygotowany przez rozpuszczenie 6.36 g. Na2CO3 (bezwodny) i 3,36 g. NaHCO3 w wodzie destylowanej i rozcieńczony do objetości 1 litra [9].
3)Bufor (pH 4,9).- 0,2 M octan sodowy (6,5 NaOAc : 3,5 HOAc) przygotowany przez dodanie 65 ml 0,2 M octanu sodu (27,2 g. C2H3O2Na.3H2O na litr) do 35 ml. 0,2 M kwasu octowego (11,3 ml czystego lodowatego kwasu octowego na litr).
4)Kwas trichlorooctowy: 7% i 20%
5)Molibdenian- 5g molibdenianu amonu rozpuścić w ok. 5M roztworze H2SO4 przeygotowanym przez dodanie 14 ml stężonego roztworu kwasu siarkowego do 86ml wody destylowanej (kwas dodawać powoli, ciągle meiszając)
6)Kwas aminonaftolosulfonowy (odczynnik redukujący): 1:2:4-kwas aminonaftolosulfonowy (0,2 g) 12 g pirosiarczynu sodu oraz 2,4g krystaliczny siarczan sodowy (Na2 SO3 .7H2O) należy rozpuścić w 100 ml wody destylowanej.
7)Chlorek cyny (odczynnik redukujący): 10g chlorku cyny (SnCl2) rozpuścić w 100 ml ok. 5M HCl (w stosunku 1:1)- roztwór ten jest zdatny do użytku przez miesiąc (nalezy go przechowywać an lodzie).Odczynnik redukujący należy przygotować w rozcieńczeniu 1 do 20 (w 5M H2SO4)- tak przygotowana odczynnik jest stabilny 1 dzień.
8)Stock - fosforan (standard): 2,194 g diwodorofosforanu potasu należy rozpuścić w 500 ml wody destylowanej, całość dokładnie wymieszać. Do roztworu dodać kilka kropli chloroformu w celu zabezpieczenia przed wzrostem mikroorganizmów. Roztwór ten zawiera 1mg fosforu na mililitr [9].
9)Przygotowanie standardu fosforu ze stocku (0,004 mg P w 1 ml): 2 ml fosforanu (ze stocku) odmierzyć do kolby o poj. 500 ml i uzupełnić do kreski za pomocą wody destylowanej, całość dokładnie wymieszać. Zakonserwować roztwór dodając kilka kropli chloroformu [9].
Wykonanie:
Do probówki odpipetowano po 3 ml buforu i substratu, próbkę ogrzewano w łaźni wodnej w 37°C przez 3-4 minut. Do próbki dodano 0,3 ml osocza lub surowicy, całość łagodnie wytrząśnięto i pozostawiono w inkubacji przez kolejne 15 minut. Po upłyiwe inkubacji, do próbki dodano 1,2 ml 20% kwasu trichlorooctoweg- zatrzymanie hydrolizy. Całość wymieszano i zwirowano w celu usunięcia białek (próbka może być rónież przefiltrowana) [9].
Kontrola: 0,3 ml osocza dodano do 6 ml wody, wymieszano, po czym próbkę zmieszano z 1,2 ml 20% kwasu trichlorooctowego, całość przefiltrowano.
Blank (próba zerowa)- do 3 ml buforu dodano 3 ml substratu, 0,3 ml wody oraz 1,2 ml 20% kwasu trichlorooctowego [9].
Oznaczanie fosforu:
Do 5 ml każdej z próbek (testowej, kontrolnej i blanka oraz próbki ze standardowym roztworem fosforu (0,02 mg P) , dodano 0,8 ml molibdenianu oraz 0,2 ml kwasu aminonaftolosulfonowego. Próbki wymieszano i pozostawiono na 10 minut w celu pojawienai się niebieskiego koloru. Następnie, w próbkach zmierzono absorbancję (ekstynkcję) za pomocą foto-elektrycznego kolorymetru, wyposażonego w filtr czerwonego światła.
Równomolowe ilości fenolu i fosforanu są uwalniane w trakcie hydrolizy fenylofosforanu. Dla każdej czasteczki fenolu o masie molowej równej 94, 11 g/mol, przypada cząsteczka fosforanu zawierającego jeden atom fosforu (P)o masie molowej równej 31 g/mol. Tak więc, około trzy razy więcej fenolu (wagowo) uwalniane jest w określonym czasie jako fosforan wyrażony jako P.
Jednostak Kinga-Armstronga zdefiniowana jest jako ilość enzymu,która uwalnia 1 mg fenolu z fenylofosforanu. Jednostka ta możne być również zdefiniowana jako ta, która uwalnia 1/3 mg P, w związku z czym konieczne jest aby pomnożyć liczbę P (mg) uwolnionego w trakcie 15 minut przez 3, aby uzyskać wartość równą jednostce Kinga-Armstrona [9].
Metoda II :
Próba testowa:
1 ml każdego buforu i substratu odpipetowano do probówki i ogrzewano w łaźni wodnej w temperaturze 37 ° C przez 3 do 4 minut. Następnie dodano 0,1 ml osocza lub surowicy, wymieszano, po czym próbkę utrzymywano w temperaturze 37°C przez 15 minut.
Dalej, dodano 2,9 ml 7% kwasu trichlorooctowego, całośc dokładnie wymieszano i odwirowano.
Kontrola: kontrolę przygotowano przez potraktowanie 2 ml wody i 0,1 ml osocza 2,9 ml 7% kwasu trichlorooctowego. Całośc wymieszano, po czym poddano filtracji
Blank: na blank skłąda sie bufor i substrat w objętośco po 1 ml oraz 0,1 ml wody i 2,9 ml 7% kwasu trichlorooctowego.
Oznaczanie fosforu:
Standard, oraz po 2 ml każdej próbki testowej, kontrolę oraz blank rozcieńczono w 3 ml wody, po czym próbki potraktowano 0,8 ml molibdenianu. Próbki dokłądnie wymieszano, po czym dodano do nich po 2 ml chlorku cyny – całośc ponownie dokładnie wymieszano. Próbki inkubowano 15 minut, a dalej odczytano otrzymane wyniki za pomocą kolorymetru, który jako wartość zero wskazuje po pomiarze próbki „blank” składającej się z 4 ml wody, 1 ml 7% kwasu trichlorooctowego, 0,8 ml molibdenianu oraz 0,2 ml chlorku cyny [9].
Obliczenia:
Otrzymane w trakcie doświadczenia wyniki pokazują, że w 2 ml próbki testowanej i kontrolnej stanowią 0,04 ml osocza, a co za tym idzie tyle samo unitów fosfatazy na 100 ml [9].
Rietz B. oraz Gullbault G.G (1975) opisali enzymatyczną metodę fluorymetryczną wykorzystywaną do określania aktywności fosfatazy alkalicznej w surowicy oraz fosfatazy kwaśnej w roztworze i na silikonowych podkładkach. Jako podłoże w doświadczeniu zastosowano fosforan 4-metylo-umbeliferonu. W roztworze, reakcja meirzona jest w temperaturze 37°C w 3-ml kuwetach [10]. Pomiar na silikonowych podkładkach również odbywa się w temperaturze 37°C, po wcześniejszym otrzymaniu stabilnych warst wszystkich wykorzystywanych odczynników przez liofilizację na powierzchni silikonowych podkładek. W metodzie wykorzystuje się jedynie ok 20 do 30 µl roztworu substratu, 50 µl roztworu buforu oraz 1 do 10 µl krwi, dzięki czemu końcowa objętość odczynnikó wynosi od 51 do 60 µl dla każdego pomiaru. W doświadczeniu mierzono szybkośc pojawienia się fluorescencyjnego 4-metyloumbeliferonu uwalnianego z fosforanu-4-metyloumbeliferonu na skutek działania enzymów, a następnie z otrzymany wartośći obliczano aktywność enzymu [10].
Umbeliferon zaliczany jest do kumaryn (7-hydroksykumaryna). Związek ten występuje w roślinach z rodzin: Umbelifere Solanaceae, Compositae i innych. Ponadto, stanowi jeden ze składników olejku eterycznego rumianku pospolitego. Zazwyczaj umbeliferon spotykany jest w wolnej postaci tzw. aglikon. Ważną cechą umbeliferonu jest zdolność do absorbcji ultrafioletowej części widma słonecznego , przez co związek ten należy do tak zwanych substancji fotochronnych. Tak więc, pochodne umbeliferonu (np.: octan umbeliferonu) znalazły zastosowanie przy produkcji kosmetyków przeciwsłonecznych [11].
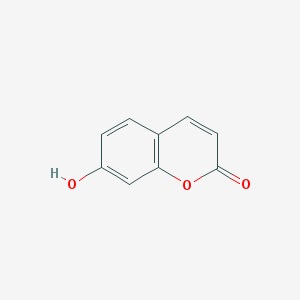
Zdjęcie: Struktura umbeliferonu, http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=5281426#itabs-2d
Dotychczas opisano wiele procedur, w których dla zwiększenia czułości fluorymetrycznej wykorzystywano związki (substraty), które są rozszczepiane enzymatycznie dając w konsekwencji produkty fluorescencyjne. Fosforan 4-metyloumbeliferonu oraz fosforan umbeliferonu wykorzystywane są np. jako substrat do oznaczania aktywności jelitowej (cielęcej) fosfatazy alkalicznej [10].
Oznaczenie aktywności fosfatazy alkalicznej wg Rietz B. oraz Gullbault G.G (1975) [10].
Odczynniki:
Wykorzystywany w doświadczeniu fluorometr poddano standaryzacji w roztworze 10 -6 mol/litr siarczanu chininy w 0,2 mol/litr H2SO4.
Fluorometryczne oznaczanie fosfatazy zasadowej [10]:
Do 0,1 ml roztworu fosforanu 4-metyloumbeliferonu dodano 2,9 ml buforu Tris. Roztwór ogrzano do temperatury równej 37 ° C przez 2 minuty w fluorymetrze).
Po 2 minutach do próbki dodano enzym, całość dokładnie wymieszano w ciepłym roztworze. Następnie, mierzono szybkość zmiany fluorescencji po upływie 1 minuty (tj. dokładnie 3 min po rozpoczęciu ogrzewania roztworu), po czym wykreślono zmiany fluorescencji (∆f / min) względem aktywności enzymatycznej [10].
Fluorometrycze oznaczanie fosfatazy kwaśnej:
Do 0,5 ml fosforanu 4-metyloumbeliferonu dodano 2,5 ml buforu cytrynianowego. Dalej postępowano zgodnie z powyższą procedurą (oznaczanie fosfatazy zasadowej). Wykres kalibracyjny ∆f/min względem aktywności enzymatycznej powinien być liniowy od 0,265 do 5,3 jednostek King-Armstrong. Na podstawie otrzymanego wykresu następnie określono aktywność enzymu w surowicy krwi [10].
P-nitrofenol jest bardzo ważnym związkeim chemicznym wykorzystywanym w biologii i medycynie. Zaliczany jest do związków wysoko-chromogennych, stabilnych będących produktem enzymatycznej katalizy wielu syntetycznych substratów. Jeden z tych syntetycznych substratów – fosforan 4-nitrofenylu (4NPP) jest wykorzystywany w chemi klinicznej do pomiaru fosfatazy alkalicznej [13].
Oznaczanie aktywności fosfatazy zasadowej oraz fosfatazy kwasowej z wykorzystaniem metody Besseya i Lowry’ego (E.Szczeklik 1963: Enzymologia kliniczna. PZWL, Warszawa, s.272).
Aktywność fosfataz podawana jest w tzw. jednostkach Besseya. Jednostka Besseya oznacza liczbę milimoli p-nitrofenolu uwolnionego w trakcie reakcji przez enzym w określonych warunkach tj.: w 1000 cm3 surowicy w trakcie 1 godziny inkubacji w warunkach metody bądź standardowych jednostkach aktywności enzymów (U/l). Jednostka Besseya oznacza taką ilość enzymu, która zdolna jest do katalizowania przemiany 1 µmola substratu w trakcie 1 minuty w temperaturez równej 30°C w optymalnych warunkach [12]. W metodzie Besseya i Lowry’ego oznaczenie fosfataz opiera się spektrofotometrycznym pomiarze stężenia p-nitrofenolu, który uwalniany jest w trakcie enzymatycznej hydrolizy p-nitrofenylofosforanu. Oznaczenie wykonuje się po zalkalizowaniu próbki badanej [12], [3].
Odczynniki wykorzystywane w trakcie oznaczenia:
1)Odczynnik A: rozpuścić 7,5 g glicyny oraz 15 mg MgCl2 w wodzie, dodać 85 ml 1M roztworu wodorotlenku sodu, po czym uzupełnić wodą do końcowej objętości równej 1000 ml (pH= ok. 10,5)
2)Odczynnik B: 0,4% p-nitrofenylofosforan disodowy w 0,001M roztworze kwasu solnego, pH= 6.5 – 8.0.
3)Odczynnik C: w celu przygotowania odczynnika C należy zmieszać równe objętości odczynnik A oraz B. 2 ml otrzymanego substratu po dodaniu 10 ml 0,02 M roztworu NaOH nie powinny wykazywać absorbancji przy 415 nm większej od 0,1 (pomiar przeprowadzony w kuwecie o grubości 1 cm). W razie potrzeby otrzymany substrat można poddać przekrystalizowaniu z dodatkiem 87% roztworu etanolu [3].
4)100 mM roztwór p-nitrofenolu: 139,1 mg w 10 ml roztworu (należy sporządzić rozcieńczenia: 1,2,4,6 mmol na 1000 ml roztworu, czyli: 1,2,4 oraz 6 mM).
Wykonanie:
Do 0,5 ml surowicu dodać 0,5 ml substratu (odczynnik C). Próbke inkubować w łaźni wodnej o temp. 37°C przez 30 minut. Po upływie czasu inkubacji próbkę należy ochłodzić w łaźni lodowej , po czym dodać do niej 5 ml 0,02 M roztworu NaOH. Zmierzyć absorbancję przy długości fali równej λ= 415 nm. Oznaczenie przeprowadzić wobec próbki kontrolnej, otrzymanej w identyczny sposób z pominięciem 30-minutowej inkubacji.
Na podstawie otrzymanych wartości, absorbancję (A) należy odczytać z krzywej aktywności w jednostkach Besseya, które oznaczają liczbę milimoli p-nitrofenolu uwolnionego przez enzym w 1000 ml surowicy, w trakcie 1- godzinnnej inkubacji w warunkach metody.
Próba wzorcowa oraz kontrolna na odczynniki otrzymywana jest w analogiczny sposób co próba badana ( z surowicą) z 0,05 ml roztworów wzorcowych lub H2O.
Do oznaczenia aktywności fosfatazy kwasowej w powyższej metodzie należy wykorzystać bufor cytrynianowy o pH= 4,8, zawierający p-nitrofenylofosforan. Próbkę składającą się z 0,2 ml surowicy, 0,35 ml H2O oraz 1 ml substratu, należy inkubować przez 30 minut w temperaturze 37°C, po upływie czasu inkubacji dodać 4 ml 0,05 M roztworu NaOH. Następnie, odczytać wartość absorbancji (A) wobec próby kontrolnej.
Wartośći fizjologiczne:
- fosfataza zasadowa: 0,8 – 2,3 j. Besseya
- fosfataza kwasowa: 0,1 – 0,6 j. Besseya [3].
Autor: Lidia Koperwas
Literatura:
[1]. Żebrowaska E., Ciereszko I., 2009. Udział kwaśnych fosfataz w gospodarce fosforanowej komórek roślinnych. Postępy Biologii Komórki, Tom 36 2009 NR 4 (583-599).
[2]. Al-saadie K., Al-Mousawi I.M., Karime N.A., 2007. Kinetics Decomposition of Some Substituted Benzendiazonium Salts in HCl Solution. National Journal of Chemistry, 2007, Volume 25, 195-205.
[3]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s.556-559.
[4]. Making diazonium salts from phenylamine. http://www.chemguide.co.uk/organicprops/aniline/makediazo.html
[5]. http://exceldiag.com/catalogs/10003.pdf
[6]. Wendt U., Polek A., Łangowski K., Rogulski J., 2009. Jednostki układu SI i ich zastosowanie w medycynie laboratoryjnej. diagnostyka laboratoryjna Journal of Laboratory Diagnostics 2009 • Volume 45 • Number 2 • 155-162. http://diagnostykalaboratoryjna.eu/journal/DL-2_2009._str_155-162.pdf
[7]. M/s Excel Diagnostics Pvt. Ltd. Plot NO. 89, Road No.8, ALEAP I.E., Near Pragathi Nagar, Opp. Kukatpally JNTU, Hyderabad - 500 090 (A.P.) INDIA. http://exceldiag.com/catalogs/10003.pdf
[8]. http://www.thermo.com/eThermo/CMA/PDFs/Various/File_28255.pdf
[9]. King E.J., Abul-Fadl M.A.M., Walker P.G., 1951. KING-ARMSTRONG PHOSPHATASE ESTIMATION
BY THE DETERMINATION OF LIBERATED PHOSPHATE. J. clin. Path. (1951), 4, 85. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1023361/?page=1
[10]. Rietz B., Gullbault G.G., (1975). Fluorometric Assay of Serum Acid or Alkaline Phosphatase , Either in Solution or on a Semisolid Surface. CLIN. CHEM.21/12, 1791-1794 (1975). http://www.clinchem.org/content/21/12/1791.full.pdf
[11]. http://www.farmakognozja.farmacja.pl/fitochem/index.php?grupa=kumaryny&strona=3
[12]. http://analiza.ovh.org/cw/cw3.pdf
[13]. http://www.wiener-lab.com.ar/wiener/catalogo/archivos/13498_alp405aa_liquida_pl.pdf
[14]. Bowers G.N., Mc Comb R.B., Christensen R.G., Schaffer R., 1980. High-Purity 4-Nitrophenol: Purification, Characterization, and Specifications of Use as a Spectrophotometric Reference Material . Clin. Chem. 26/6 , 724-729 (1980). http://www.clinchem.org/content/26/6/724.full.pdf
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje