
|

Zamknij X
|




Oznaczanie wybranych składników surowicy
1) Chlorki
W obecności jonów srebrowych chlorki wytrącają się w postaci AgCl (biały osad). W celu przeprowadzenia doświadczenia do 0,5 ml przesączu surowicy (po odbiałczaniu kwasem trichlorooctowym) do próbki dodać 2 ml 0,2% roztworu AgNO3
2) Jony wapniowe
Wytrącanie wapnia z surowicy przeprowadza się za pomocą roztworu szczawianu amonowego. W tym celu do 2 ml surowicy należy dodać 1 ml nasyconego roztworu (COO)2(NH4)2. W wyniku reakcji obserwuje się wytrącanie białego osadu (COO)2Ca [1].
Prawidłowe wartości kwasu moczowego we krwi wynoszą 2,5 do 9mg/dl [4]. Epidemiolodzy wskazują kwas moczowy jako niezależny czynnik ryzyka zarówno chorób nerek, jak i powkłań sercowo-naczyniowych. Ponadto, jego nieprawidłowe wartości wpływają na rozwój takich chorób jak: nadciśnienie tętnicze, otyłość, choroby nerek czy cukrzycę [5].
3) Kwas moczowy
Kwas moczowy jest końcowym produktem przemiany puryn. Zaliczany jest do kwasów słabo rozpuszczalnych w płynach ustrojowych. W ciągu doby u człowieka produkowane jest około 250-750 mg kwasu moczowego, który w większości pochodzi z puryn z diety oraz z rozpadu obumierających tkanek. W większości kwas moczowy wydalany jest przez nerki (8% - 12% docierającego do nerek kwasu moczowego zostaje wydalone, zaś ok. 90% podlega tzw. zwrotnej reabsorpcji [9].
W środowisku zasadowym kwas ten powoduje redukcję odczynnika fosforowolframowego do niebiesko zabarwionej mieszaniny niższych tlenków wolframowych [1].
Przygotowanie odczynnika fosforowolframowego:
- zmieszać 50 g Na2WO4 ∙ 2H2O w kolbie z 40 ml 85% roztworu H3PO4 i 350 ml wody. Mieszaninę gotować przez 6 h pod chłodnicą zwrotną. Następnie, dodać kilka kropli bromu i ogrzewać przez kilkanaście minut bez chłodnicy zwrotnej. Po ochłodzeniu uzupełnić wodą w kolbie miarowej do 500 ml. Przechowywać w lodówce)
Wykonanie oznaczenia:
Do 4 ml przesączu po kwasie trichlorooctowym dodać kilka kropli odczynnika fosforowolframowego oraz 2 ml 20% roztworu Na2CO3. W wyniku reakcji w próbce pojawia się niebieskie zabarwienie, świadczące o obecności kwasu moczowego [1].
Kwas moczowy będący produktem metabolizmu puryn ulega kilku etapom:
Analiza składu kwasów tłuszczowych w materiale biologicznym, takim jak surowica czy tkanki, wymaga zastosowania odpowiednich metod: ekstrakcji i oczyszczania frakcji lipidowych. W badaniach najczęściej stosowaną metodą do ilościowej ekstrakcji składników lipidowych z surowicy krwi jest metoda Folcha i wsp.(1957). W celu zapobiegnięcia utlenianiu kwasów tłuszczowych w czasie ekstrakcji stosuje się odpowiednie antyutleniacze. Wśród nich najczęściej wymienia się takie jak: butylohydroksytoluen lub 2,6-di-tert-butyl-p-cresol. Co ważne, można także każdy etap przygotowania wstępnego próbek wykonywać w atmosferze azotu. Rozdział lipidów w ekstraktach z materiałów biologicznych można przeprowadzać kilkoma metodami np. za pomocą chromatografii cienkowarstwowej (ang. TLC) lub chromatografii cieczowej z użyciem kolumienek z aminopropylową fazą stałą. Chromatografia cienkowarstwowa jest czasochłonna, a dodatkowo powoduje utlenienie wielonienasyconych kwasów tłuszczowych (skrót PUFA) z powodu długotrwałej ekspozycji na powietrze. W związku z tym, większe zastosowanie znajduje rozdział na aminopropylowej fazie stałej, co pozwala na uzyskanie dokładniejszych wyników, a także znaczne skrócenie czasu całkowitej analizy [23].
4) Wykrywanie tłuszczów
Mieszanina fenolu i NaCl ( roztwór fenolu przygotowany w roztworze NaCl) powoduje wytrącanie tłuszczów surowicy. Wynikiem reakcji jest pojawiające się zmętnienie roztworu [1].
Wykonanie: do 0,5 ml surowicy dodać 4,5 ml 1% roztworu fenolu w 12% roztworze NaCl. W wyniku reakcji w probówce pojawia się zmętnienie roztworu [1].
Nadmierne stężenie kwasu moczowego może wynikać ze zwiększonej jego produkcji, jak i w przypadku upośledzonego wydalania z organizmu. Nadmierna produkcja kwasu moczowego może być związana np. ze stosowaniem diety bogatobiałkowej, spożywaniem alkoholu lub defektem enzymatycznym szlaku metabolizmu puryn. U osób z upośledzoną filtracją, w przebiegu choroby nerek, stężenie kwasu moczowego zależy w dużej mierze od wydolności jelitowego mechanizmu wydalniczego. Przeprowadzone badania wykazały, że wyższe stężenie kwasu moczowego odnotowuje się u mężczyzn oraz u kobiet po menopauzie z uwagi na urykozuryczne działanie hormonów – estrogenów [10].
Funkcje ochronne kwasu moczowego
Przeprowadzone badania wykazały, że kwas moczowy spełnia również kilka ważnych funkcji w organizmie, m.in. : ma on działanie antyoksydacyjne. Ponadto, większenie jego stężenia w surowicy może wykazywać także korzystne działanie w chorobach neurodegeneracyjnych. Co więcej, zaobserwowano poprawę funkcji śródbłonka u pacjentów ze zdiagnozowaną niewydolnością serca, u których udało się obniżyć stężenie kwasu moczowego za pomocą inhibitora oksydazy ksantynowej (allopurinolu) [10].
Kwas moczowy spełnia także ważne funkcje fizjologiczne - udowodniono jego działanie antyoksydacyjne, kiedy to związek ten może odgrwać rolę neuroprotekcyjną w chorobach ośrodkowego układu nerwowego (OUN), w tym w stwardnieniu rozsianym czy chorobie Parkinsona . Dodatkowo, może uczestniczyć w pobudzaniu wrodzonych mechanizmów odpornościowych, oddziałując na limfocyty T oraz komórki dendrytyczne [10]. Wykazano również działanie prozapalne kwasu moczowego, gdyż pobudza on syntezę cytokin: czynnika chemotaktycznego monocytów 1 (MCP-1), interleukin 1β i 6 (IL-1β, IL-6) oraz czynnika martwicy nowotworów alfa (TNFα) [10].
Mocznik (diamid kwasu węglowego) jest związkiem bardziej zasadowym niż monoamidy kwasów karboksylowych. Tworzy on łatwo sole z mocnymi kwasami np. wytrącanie mocznika z roztworów wodnych pod wpływem kwasu azotowego (V). W wyniku tej reakcji powstaje trudno rozpuszczalny azotan (V) mocznika. W trakcje ogrzewania mocznika powstaje dimocznik (biuret) a całej reakcji towarzyszy wydzielanie amoniaku. Biuret w środowisku zasadowym występuje w formie enolowej. W reakcji z jonami Ca2+ tworzy związki kompleksowe, charakteryzujące się fioletowym zabarwieniem. Mocznik (podobnie jak aminy alfatyczne I-rzędowe) ulega reakcji z kwasem azotowym (III), w wyniku czego wydziela się gazowy azot, a sam mocznik ulega degradacji do dwutlenkuwęgla i wody [18].
Reakcja mocznika z kwasem azotowym (V)
W reakcji mocznika z kwasem azotowym (V) powstaje trudno rozpuszczalny azotan (V) mocznika.
Wykonanie:
Kroplę stężonego roztworu mocznika należy umieścić na szkiełku podstawowym i dodać do niej kroplę stężonego roztworu kwasu azotowego (HNO3). Po chwili w wyniku reakcji tworzą się kryształy, które należy oglądać pod mikroskopem [18].
Odczyn Piotrowskiego - odczyn biuretowy
Wykonanie:
Około 100 g mocznika należy ogrzewać w suchej probówce do momentu, aż z próbki zacznie się wydzielać amoniak. Próbkę ostudzić, a powstały biuret rozpuścić w roztworze wodorotlenku sodu (NaOH). Nastęnie, do próbki dodać ok. 3-4 kropli rozcieńczonego roztworu CuSO4. W wyniku reakcji w próbce powstaje fioletowe zabarwienie roztworu [18].
Reakcja mocznika z kwasem azotowym (III)
Wykonanie:
Do 1 ml stężonego roztworu mocznika dodać ok. 3-4 kropli roztworu azotanu (III) sodu oraz 1 ml rozcieńczonegoroztworu kwasu siarkowego (lub solnego). W wyniku reakcji dochodzi do rozkładu mocznika z wydzieleniem azotu [18].
Metoda ureazowa do oznaczania mocznika w surowicy krwi [1].
Ureaza rozkłąda mocznik na amoniak. Po etapie strącenia białek do próbki dodaje się odczynnik Nisslera, który w reakcji z amoniakiem daje żółte zabarwienie. Jest to wynikiem wytworzenia się jodku amidoksyrtęciowego (II)- w środowisku zasadowym [1].
Roztwór wzorcowy: odczynnik Nisslera
W kolbie należy rozpuścić 30 g Kiw 20 ml wody, po czym dodać 22 g jodu i 30 g metalicznej rtęci. Roztwór dokładnie wytrząsać przez 10 minut, aż do momentu zmiany zabrawienia ( ciemna barwa roztworu zmieni się w jasną). Ciepły roztwór należy ochłodzić pod bieżącą wodą, po czym dalej wytrząsąć, aż do momentu pojawienia się zielonej barwy. Na tym etapie, zdekantować ciecz znad nierozpuszczonej rtęci,pozostałą rtęć przepłukać kilkakrotnie wodą, a na koniec dołączyć ją do zdekantowanej cieczy. Roztwór uzupełnić wodą do końcowej objętości równej 400 ml, a całość przesączyć [1].
Roztwór roboczy: odczynnik Nisslera
Do 75 ml roztworu wzorcowego należy dodać 75 ml wody i 350 ml 10% roztworu wodorotlenku sodu. Mieszaninę inkubować kilka dni w temp. pokojowej, aż do momentu wydzielenia się rdzawo-brunatnego osadu w próbce. Do oznaczeń należy zawsze używać tylko klarownego supernatantu, którego po zmętnieniu nie należy przesączać, a jedynie odwirować. Otrzymany odczynnik należy przechowywać w ciemnej butelce [1].
Wykonanie oznaczenia: do 3,2 ml 3% roztworu Na2SO4 dodać 0,2 ml krwi oraz 20 mg ureazy. Probówkę zamknąć korkiem, dokładnie wymieszać i inkubować w łaźni wodnej przez 20 minut w temp. 37°C. Po upływie czasu inkubacji w celu wytrącenia białek do próbki dodać: 0,3 ml 10% roztworu ZnSO4(10% roztwór ZnSO4 ∙7H2O) i 0,3 ml 0,5M roztworu NaOH. Próbkę dokałdnie wytrząsnąć, a następnie odwirować. Po wirowaniu pobrać 2 ml otrzymanego supernatantu, dodać do niego 5 ml wody i 1 ml roztworu roboczego odczynnika Nesslera. W drugiej probówce sporządzić roztwór wzorocwy : zmieszać 2 lub 5 ml roztworu NH4Cl (tj.: 0,01 mg N w 1 ml (25 ml roztworu NH4Cl o stężeniu 1,53mg/ml + 10 ml H2SO4 uzupełnić wodą do obj. 1000 ml)), 2 lub 5 ml wody oraz 1 ml odczynnika Nesslera. Zmierzyć wartość absorbancji wobec próby kontrolnej przy długości fali równej 430 nm. Na podstawie otrzymanych wyników onliczyć ilośc mg mocznika w 100 ml, wg wzoru:
mg mocznika/ 100 ml= Apb / Aw x cw x 100/0,1 x 2,14
gdzie: cw = liczba mg N we wzorcu (0.02 mg przy 2 ml lub 0,05 mg przy 5 ml wzorca). 1 mg azotu odpowiada 2,14 mg mocznika [1].
Metoda z diacetylomonooksymem [1].
W warunkach podwyższonej temperatury i w środowisku kwasowym dochodzi do kondensacji mocznika z diacetylomonooksymem. W wyniku reakcji powstaje związek o żółtym zabarwieniu. W wyniku kondensacji wytwarza się także hydroksylamina, która hamuje tę reakcję. Dodanie antypiryny, która łączy się z hydroksylaminą powoduje zniesienie jej niekorzystnego wpływu. Natężenie barwy w roztworze jest wprost proporcjonalne do stężenia mocznika w badanej próbce.
Wykonanie: do 2 ml odczynnika roboczego (tj.: rozpuścić 0,2 g antypiryny i 0,25 g diacetylmonooksymu w roztworze zawierającym 50 ml 36% roztworu kwasu H2SO4, 10 ml 25% kwasu octowego i 3 ml etanolu, całośc uzupełnić do obj. 100 ml) dodać 0,04 ml rozcieńczonej surowicy lub moczu (próba badana). Roztwór dokłądnie wymieszać, po czym przykryć folią aluminiową i inkubować we wrzącej łąźni wodnej (15 minut). Próbkę ochłodzić, a następnie zmierzyć wartość absorbancji próby badanej i wzorcowej względem próby konrolnej. Oznaczenie wykonywać przy λ=460 nm. Intensywnośc zabarwienia nie zmienia się przez 30 minut. Na podsawie otrzymanych wyników obliczyć:
mg/100 ml = Apb / Aw x 5 mg/100 ml x 0,04 ml/0,002 ml = Apb / Aw x 5 x 20= Apb / Aw x 100
gdzie: 0,04 ml 20-krotnie rozcieńczonej surowicy = 0,002 ml surowicy nierozcieńczonej)
mmol/l= Apb / Aw x 100mg x10/ 60 mg [masa milimolowa] = Apb / Aw x 16,66
W przypadku obliczeń stężenia w moczu należy uwzględnić jego 100-krotne rozcieńczenie, a otrzymane wyniki uzyskane wg powyższych wzorów należy pomnożyc przez 5. W przypadku surowicy jej rozcieńczenie jest 20-krotne [1].
Oznaczanie mocznika kinetyczną metodą hydrolizy enzymatycznej [11].
Jony amonowe NH4+ będące produktem rozpadu białek, wykorzystywane są w biosyntezie związków azotowych, zaś ich nadmiar wydalany jest w postaci mocznika. Mocznik produkowany jest w wątrobie w tzw. cyklu mocznikowym [11].
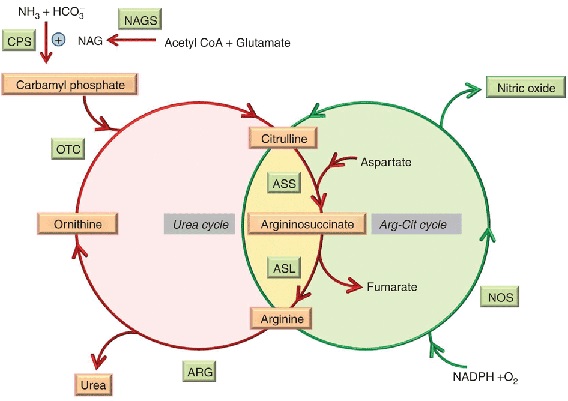
Zdjęcie: Schemat cyklu mocznikowego, http://www.nature.com/gim/journal/v15/n4/fig_tab/gim2012166f3.html [12].
W badanich krwi, zawartość mocznika zazwyczaj wyrażana jest jako azot mocznikowy krwi, określany skrótem BUN (ang. Blood Urea Nitrogen). W warunkach fizjologicznych wartość ta stanowi 50% azotu niebiałkowego, zaś zawartość mocznika kształtuje się na poziomie od 15mg do 40 mg/100ml (2.49-6.64 mmol/l) [11].
Obserwowany podwyższony poziom mocznika we krwi najczęściej kojarzony jest z chorobami nerek, aczkolwiek może to być również wynik odwodnienia organizmu, czy stosowania diety wysokobiałkowej. Z kolei, powodem obniżonego stężenia mocznika we krwi może być dieta niskobiałkowa lub głodzenie. Do wyników takich przyczynia sie także nadmierne nawodnienie (spożywanie zbyt dużych ilości płynów), a także wspomniane już uszkodzenia wątroby [11].
Stosowane do oznaczania mocznika metody hydrolizy enzymatycznej opierają się na reakcjach katalizowanych przez ureazę (odpowiedzialną za rozkład mocznika na amoniak i dwutlenek węgla), gdzie mierzone jest stężenie produktów reakcji hydrolizy mocznika, wg równania:
Enzym ureaza
CO(NH2)2 + 3H2O --------------------> 2NH4+ + CO2 + 2OH-
Mocznik
Ureaza katalizuje hydrolize mocznika, w wyniku czego powstaje amoniak, który następnie z 2-oksoglutaranem w obecności zredukowanej formy dinukleotydu nikotynoamidoadeninowego (NADH) pod wpływem dehydrogenazy glutaminianowej ulega przekształceniu do L-glutaminianu. W metodzie mierzone jest pasmo przy wartość absorbancji równej 340 nm – pochodzace od NADH [11].
Wykonanie oznaczenia:
a) Przygotowanie próby odczynnikowej (kontrola)
Do probówki typu eppendorf należy odmierzyć 1 ml odczynnika do oznaczeń 1-UREA, próbkę ogrzać do temperatury oznaczenia ok. 25-30°C, a następnie dodać do niej 250 μl odczynnika do oznaczeń 2-UREA. Całośc dokładnie wymieszać, po czym przenieść 800 ml roztworu do kuwety. Wykonać pomiary absorbancji A1 i A2 próby wzorcowej i badanej względem próby odczynnikowej przy długości fali równej λ= 340 nm [11].
b) Przygotowanie próby wzorcowaej
Do probówki typu eppendorf odmierzyć 1 ml odczynnika do oznaczeń 1-UREA. Próbkę ogrzać do temperatury oznaczenia tj. 25-30°C. Następnie, dodać 10 μl roztworu wzorcowego, całość dokładnie wymieszać > Próbkę inkubować przez ok. 5 min. w temperaturze pokojowej. Po upływie czasu inkubacji dodać 250 μl odczynnika do oznaczeń 2-UREA. Dokładnie wymieszać, po czym przenieść 800 ml roztworu do kuwety. Po upływie ok. 1 min. od dodania odczynnika należy odczytać absorbancję A1 próby wzorcowej względem próby odczynnikowej przy długości fali równej 340 nm. Następnie, nie wyjmując kuwety (dokładnie po upływie 1 min.) należy zmierzyć wartość absorbancji A2 próby wzorcowej względem próby odczynnikowej. Każde oznaczenie wykonać w trzech powtórzeniach. Na podstawie otrzymanych wartości należy obliczyć wartość zmiany absorbancji na minutę: ∆A/min. (A1 – A2) [11].
c) Przygotowanie próby badanej
Do probówki typu eppendorf należy odmierzyć 1 ml odczynnika do oznaczeń 1-UREA. Próbkę ogrzać do temperatury oznaczenia 25-30°C, po czym dodać 10 μl roztworu wzorcowego. Dokładnie wymieszać i inkubować 5 min, a następnie dodać 250 μl odczynnika do oznaczeń 2-UREA. Całą zawartość probówki dokładnie wymieszać, po czym przenieść 800 ml roztworu do kuwety pomiarowej.Próbkę inkubować ok. 1 min.(od momentu dodania odczynnika) i odczytać absorbancję A1 próby badanej względem próby odczynnikowej przy długości fali λ= 340 nm. Następnie, nie wyjmując kuwety, dokładnie po upływie 1 min. należy dokonać pomiaru absorbancji A2 próby badanej względem próby odczynnikowej- oznaczenia powtórzyć trzy razy,a na koniec obliczyć zmianę absorbancji ∆A/min. (A1 –A2) (jak wyżej) [11].
Na podstawie otrzymanych wyników obliczyć stężenie mocznika w badanej próbie korzystając ze wzoru:
Stężenie mocznika = ∆A(PB) / ∆A(PW) x stężenie wzorca
Gdzie:
- 1 mg mocznika odpowiada 0,467 mg azotu mocznikowego (BUN) [11].
Zastosowanie metody enzymatycznej do oznaczania kwasu moczowego (wg. E.Praetorius, H.Polusen 1953: J.Clin.Lab.Invest., 5:273-279) [1].
Jako materiał badawczy w metodzie tej można wykorzystać surowicę, osocze heparynowe lub 10-krotnie rozcieńczony mocz. Jako wzorzec wykorzystywany jest kwas moczowy o stężeniu 4 mg/100ml(0,24mmol/l).
Wykonanie:
- Przygotowanie próby badanej: do 1 ml odczynnika roboczego (tj.: 40µmol 4-aminoantypiryny, 0,2mmol DHBS(kwas 3,5-dichloro-2-hydroksybenzenosulfonowy), 18U urykazy oraz 500U peroksydazy rozpuścić w 100 ml wody, delikatnie wymieszać) należy dodać 50 µl surowicy
- Przygotowanie próby wzorcowej: do 1 ml odczynnika roboczego dodać 50 µl wzorca
- Przygotowanie próby ślepej: 1 ml odczynnika roboczego z dodatkiem 50µl tego odczynnika [1].
Próbki dokładnie wymieszać i inkubować w temperaturze 25°C przez 10 minut. Po upływie inkubacji odczytać wartość absorbancji (A) dla próby badanej i wzorcowej względem próby ślepej przy długośi fali równej λ=500 nm. Nastęnie, obliczyć stężenie kwasu moczowego, uwzględniająć stężenie wzorca równe 4 mg / 100 ml, wg wzoru:
mg /100 ml = (Apb /Aw) x 4 mg /100 ml [1].
Pomiar stężenia kwasu moczowego z wykorzystaniem gotowego zestawu odczynników [procedura z zestawu firmy BioSystems ze strony: http://chemklin.sum.edu.pl/uploaded/Chemia%20kliniczna/Kwas%20moczowy.pdf)
W wyniku sprzężonych reakcji kwas moczowy w próbie badanej tworzy barwny kompleks. Dzięki temu jego stężenie może być mierzone z wykorzystaniem spektrofotometru.
Na rynku dostępne są gotowe zestawy do pomiaru stężenia kwasu moczowego, których działanie opiera się na reakcjach enzymatycznych (wykorzystanie specyficznych enzymów, które reagują z kwasem moczowym: urykaza, peroksydaza) [13].
Materiał do badań:
Suroica, osocze lub mocz należy pobrać zgodniez panującymi procedurami, a przed samym wykonaniem oznaczenia (w przypadku próbki moczu) nalęzy ją rozcieńczyć wodą destylowaną w stosunku 1:10. Stabilność kwasu moczowego w surowicy wynosi 7 dni w temperaturze od 2°C do 8°C. W moczu stabilnośc kwasu moczowego wynosi 4 dni w temperaturze pokojowej, jeżeli pH moczu doprowadzone jest do >8 przez dodanie roztworu NaOH [13].
Odczynnik zestawu:
- odczynnik (A): fosforan 100mmol/l, detergent 1,5 g/l, dwuchlorofenylosulfonian 4mmol/l, urykaza>0,12 U/ml, oksydaza askorbinowa >5U/ml, peroksydaza >1U/ml, 4-aminoantypiryna 0,5mmol/l, pH=7.8.
- standard (S) kwasu moczowego: kwas moczowy 6mg/dl (357µmol/l) [13].
Wykonanie:
- Odczynniki należy doprowadzić do temperatury pokojowej
- Do odpowiednio podpisanych probówek (tj. próba ślepa, standard, próba badana) odpipetować :
1) próba ślepa: 25 µl wody destylowanej oraz 1 ml odczynnika (A)
2) standard: 25 µl standardu (S) kwasu moczowego oraz 1 ml odczynnika (A)
3) próba badana: 25 µl próby badanej i 1 ml odczynnika (A)
- Zawartość probówek dokładnie wymieszać, po czym inkubować probówki przez 10 minut w temperaturze pokojowej (w granicach 16- 25°C) lub przez 5 minut w teperaturze 37°C.
- Dokonać pomiaru absorbancji (A) standardu i próby badanej przy długości fali równej 520 nm wobec próby ślepej. Powstające zabarwienie w próbkach jest stabilne przez ok. 30 minut [13].
Na podstawie otrzymanych wyników należy obliczyć stężenie kwasu moczowego w próbie wg wzoru :
A próby/ Astandardu x C standardu x wspólczynnik rozcieńczenia próby = Cpróby[13].
Oznaczanie kwasu moczowego w surowicy krwi (procedura z: Metody badań analitycznych. PZWL, Warszawa 1959, instr.30) [1].
Kwas moczowy w reakcji z węglanem sodu powoduje redukcję kwasu fosforowolframowego do błękitu wolframowego. Natężenie powstałego w reakcji zabarwienia jest proporcjonalne do stężenia kwasu moczowego w badanej próbce. Na oznaczanie kwasu moczowego w danej próbce mają wpływ (interferują):
Hemoliza, zwiększone stężenie kwasu askorbinowego, niektóre leki np. kofeina, salicylany, fenacetyna [1].
Wykonanie: 1 ml surowicy zmieszać z 1 ml 20% roztworu TCA (Cl3COOH) i 3 ml wody. Dokładnei wymieszać, a po upływie 10 minut roztwór przesączyć. Do 2 ml przesączu dodać 0,1 ml odczynnika fosforowolframowego oraz 0,9 ml 22% roztworu Na2CO3. Inkubować 10 min, a następnie zmierzyć wartość absorbancji przy długości fali równej 610 nm wobec wody [1].
Przygotowanie próby wzorcowej: do 1 ml roztworu wzorcowego kwasu moczowego (stężenie 2 mg/100 ml) dodać 0,4 ml TCA, 0,6 ml roztworu mocznika (40%), 0,1 ml odczynnika fosforowolframowego oraz 0,9 ml roztworu Na2CO3. Inkubować 10 minut po czym zmierzyć absorbancję przy λ=610 nm wobec wody [1].
Na podstawie uzyskanych wynikó oblicza się stężenie kwasu moczowego w surowicy wg wzorów:
mg/100 ml = Apb / Aw x 2 mg/100ml x 1 ml wzorca/0,4 ml surowicy = Apb / Aw x 5 mg/100 ml lub
µmol/l = (Apb / Aw) x 297, 4 µmol/l
Zależność absorbancji od stężenia kwasu moczowego w badanej próbce ma przebieg prostolinijny w granicach do 10mg/100 ml [1].
Oznaczanie żelaza w surowicy krwi
Żelazo dwuwartościowe w reakcji z 2,2’-dipirydylem tworzy związek kompleksowy o różowym zabarwieniu. Z kolei, żelazo dwu- i trójwartościowe w środowisku zasadowym tworzy związek wewnątrzkompleksowy z kwasem sulfosalicylowym [1].
Wykonanie:
1)Do 2 ml surowicy należy dodać 2 ml 2,5% roztworu Na2SO3 oraz 2 ml 0,1% roztworu 2,2’-dipirydylu w 3% roztworze CH3COOH. Próbke dokładnie wymieszać i inkubować we wrzącej łaźni wodnej przez 5 minut. Po upływie czasu inkubacji próbkę odwirować [1].
2)Do 3 ml surowicy dodać 3 ml 20% roztworu CCl3COOH. Dokładnie wymieszać, a po kilku minutach próbkę odwirować. Do powstałego po wirowaniu supernatantu dodać kilka kropli 20% roztworu kwasu sulfosalicylowego i 1 ml stężonego roztworu NH3 – dodawać do momentu pojawienia się w próbce żółtego zabarwienia [1].
Żelazo występuje w organizmie w różnych formach, m. in. w hemoglobinie, jako żelazo tkankowe, a także w mioglobinie. Żelazo przenoszone jest przez białkowy nośnik zwany apotransferyną, a kompleks białka z żelazem określany jest mianem transferyny. Ferrytyna bedąca białkiem kompleksującym jony żelaza, jest obecna w większości komórek, a dodatkowo stanowi rezerwuar żelaza dostępny dla tworzenia hemoglobiny i innych białek, które zawierają grupę hemową. Ferrytyna i transferyna są związkami obecnymi w komórkach błony śluzowej jelit, razem biorą udział w regulacji wchłaniania [24].
Główne zaburzenia metaboliczne są związane z niedoborem lub nadmiarem żelaza w organizmie. Zmiany w poziomie żelaza obserwuje się w wielu różnych zaburzeniach jak np.: astma, choroby sercowo-naczyniowe, przewlekle zapalenie wątroby czy choroby nerek. W badaniach najczęściej diagnozowanym zaburzeniem jest niedokrwistość z niedoboru żelaza, która zazwyczaj występuje u dzieci, kobiet w ciąży i starszych osób. Ponadto, do niedikrwistości mogą prowadzić takie choroby jak wrzody żołądka, wrzody dwunastnicy czy rak żołądka. Nadmiar żelaza diagnozuje się np. w takich zaburzeniach jak: hemosyderoza, hemochromatoza i anemia sideroblastyczna [24].
W standardowych metodach analitycznych, związki redukujące takie jak np. hydroksyloamina są niezbędne do konwersji żelaza (III) w żelazo (II) z wytworzeniem czerwono-pomarańczowego kompleksu z o-fenantroliną. Jako związek silnie trujący (mutagen) , hydroksylamina wywołuje niepożądane efekty zdrowotne związane z jej absorpcją przez skórę. Tak więc, związek ten działa drażniąco na skórę, oczy, błony śluzowe i drogi oddechowe. Jako alternatywę do badań wybrano kwas askorbinowy, który również wykazuje silne właściwości redukujące, a w porównaniu do hydroksylaminy jest on przyjazny dla środowiska [25]. Przeprowadzone badania wykazały, że kwas askorbinowy jako prosty, tani i przyjazny środowisku środek redukujący, wykazuje maksimum zdolności redukujących żelazo (III) w stężeniu poniżej 4,46 x 10-4M, w pH roztworu 1-3 [25].
Autor: Lidia Koperwas
Literatura:
[1]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s.580-582, 592-595.
[2]. http://www.ibmb.uni.wroc.pl/studia/2.pdf
[3]. Rodriguez-Vico F., Martinez-Cayuela M., Zafra M.F., Garcia-Peregrin E., Ramirez H., 1991. Lipids 26 (77-80).
[4]. Niebiałkowe związki azotowe -mocznik, kwas moczowy, kreatynina; ilościowe oznaczanie kreatyniny metodą Folina-Wu. Ćwiczenie nr 13. http://farmacja.cm-uj.krakow.pl/public_includes/upload/bioch_farm_cwiczenie_nr_13.pdf
[5]. Posadzy-Małaczyńska A., Tykarski A., 2011. Kwas moczowy w chorobach sercowo-naczyniowych – co nowego?. Borgis - Postępy Nauk Medycznych s3/2011, s. 46-50. http://www.pnmedycznych.pl/shown.php?ktory=4061
[6]. Drabczyk R., 2014. Kreatynina. Medycyna praktyczna dla pacjentów. http://nefrologia.mp.pl/diagnostyka/show.html?id=51973
[7]. http://www.merckmillipore.com/PL/en/product/Creatinine,MDA_CHEM-105206
[8]. http://www.merckmillipore.com/INTL/en/product/poland/chemicals/mocznik,MDA_CHEM-108486
[9]. Majdan M., Borys O., 2010. DNA i schorzenia towarzyszące podwyższonemu stężeniu kwasu moczowego. ANNALES ACADEMIAE MEDICAE STETINENSIS ROCZNIKI POMORSKIEJ AKADEMII MEDYCZNEJ W SZCZECINIE 2010, 56, SUPPL. 1, 34–39. http://www.pum.edu.pl/__data/assets/file/0009/29817/SUPLEMENT_56-01_05.pdf
[10]. Januszkiewicz-Caulier J., Franek E., 2011. Kwas moczowy w chorobach nerek, serca i naczyń. 31
K L I N I C Z N A I N T E R P R E T A C J A W Y N I K Ó W B A D A Ń. Choroby Serca i Naczyń 2011, tom 8, nr 1, 31–37. http://czasopisma.viamedica.pl/chsin/article/viewFile/18633/14657
[11]. Oznaczanie mocznika w płynach ustrojowych metodą hydrolizy enzymatycznej, http://pchba.amu.edu.pl/cw%20CBA/cw3.pdf
[12]. Ayelet Erez MD, PhD, 2013. Argininosuccinic aciduria: from a monogenic to a complex disorder. Genetics in Medicine (2013) 15, 251–257, http://www.nature.com/gim/journal/v15/n4/fig_tab/gim2012166f3.html.
[13]. http://chemklin.sum.edu.pl/uploaded/Chemia%20kliniczna/Kwas%20moczowy.pdf
[14]. Hunter G., 1957. A method for deproteinization of blood and other body fluids. J.clin. Path. (1957), 10, 161.
[15]. Sakuma R., Nishina T., Kitamura M., 1987. Deproteinizing Methods Evaluated for Determination of Uric Acid in Serum by Reversed –Phase Liquid Chromatography with Ultraviolet Detection. Clin. Chem. 33 No 8, 1427-1430 (1987). [16]. Daykin CA, Foxall PJ, Connor SC, Lindon JC, Nicholson JK. The comparison of plasma deproteinization methods for the detection of low-molecular-weight metabolites by (1)H nuclear magnetic resonance spectroscopy. Anal Biochem. 2002 May 15;304(2):220-30. http://www.ncbi.nlm.nih.gov/pubmed/12009699
[17]. https://www.caymanchem.com/pdfs/700460.pdf
[18]. http://usfiles.us.szc.pl/pliki/plik_1262018104.pdf
[19]. http://www.wiener-lab.com/wiener/catalogo/archivos/6322_creatinina_enzimatica_aa_liquida_pl.pdf
[20]. http://www.diag.pl/Badanie-Klirens-kreatyniny.85+M51588beb2ff.0.html
[21]. Fossati P., Prencipe L., 2010. Chromogenic System for Measuring Hydrogen Peroxide: The Enzymatic Uric Acid Assay. Clinical Chemistry May 2010 vol. 56 no. 5 865-866. http://www.clinchem.org/content/56/5/865.full
[22]. http://chemklin.sum.edu.pl/uploaded/kreatynina.pdf
[23]. Bugajska J., Berska J., Hodorowicz-Zaniewska D., Sztefko K., 2010. Walidacja metody oznaczania kwasów tłuszczowych frakcji fosfolipidów w surowicy krwi. diagnostyka laboratoryjna, Journal of Laboratory Diagnostics 2010 • Volume 46 • Number 2 • 125-130. http://diagnostykalaboratoryjna.eu/journal/DL_2_2010._str_125-130.pdf
[24]. Bezpośrednia metoda kolorymetryczna do oznaczania żelaza w surowicy krwi lub osoczu. Fer-color, Wiener-lab. http://www.wiener-lab.com/wiener/catalogo/archivos/12055_fer_color_aa_liquida_pl.pdf
[25]. Elmagirbi A., Sulistyarti H., Atikah, . Study of Ascorbic Acid as Iron (III) Reducing Agent for Spectrophotometric Iron Speciation. J. Pure App. Chem. Res., 2012, 1(1), 11-17. http://jpacr.ub.ac.id/index.php/jpacr/article/view/101/98
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje