
|

Zamknij X
|



W ludzkim mózgu znajduje się około 1011 komórek nerwowych, z których każda z nich może być połączona z wieloma innymi komórkami. Komórki nerwowe są zgrupowane w ośrodkowym i obwodowym układzie nerwowym i odpowiadają za szybkie przekazywanie informacji w organizmie. Narząd ten jest odpowiedzialny nie tylko za naszą pamięć i myślenie, ale również scala informacje z zewnątrz umożliwiając w ten sposób dostrzeganie osób, przedmiotów czy zdarzeń. Wywołuje on także reakcję na te zdarzenia na przykład na ruch bądź inne działania oraz odpowiada za nasze zachowania społeczne [1].
W miejscach, gdzie komórki nerwowe lub ich wypustki nie są z sobą zespolone, lecz stykają się i przekazują sobie bodźce, znajdują się połączenia synaptyczne (synapsy). Każda synapsa składa się z dwóch części - presynaptycznej oraz postsynaptycznej, oddzielonych od siebie wąską szczeliną synaptyczną. Część presynaptyczna zawiera pęcherzyki ze specjalną substancją chemiczną – neuroprzekaźnikiem (np. acetylocholina, peptydy, noradrenalina), natomiast w błonie części postsynaptycznej znajdują się receptory dla neuroprzekaźników. Informacja z części presynaptycznej na postsynaptyczną jest przekazywana za pośrednictwem małych ilości swoistych substancji chemicznych zwanych neurotransmiterami (neuroprzekaźnikami). Produkowanie neuroprzekaźników następuje w neuronach, a ich magazynowane w pęcherzykach presynaptycznych, z których są uwalniane pod wpływem impulsu depolaryzacyjnego. Neuroprzekaźnik jest związkiem chemicznym, którego cząsteczki przenoszą sygnały pomiędzy komórkami nerwowymi poprzez synapsy. Służy on do zmiany sygnału elektrycznego na chemiczny w synapsie i do przekazywania tego sygnału z komórki presynaptycznej do postsynaptycznej. Po przejściu przez szczelinę synaptyczną neuroprzekaźnik łączy się ze specyficznym dla niego receptorem w błonie postsynaptycznej i wywołuje odpowiednią reakcję w komórce odbiorczej [2]. Przyłączenie neuroprzekaźnika do błony postsynaptycznej powoduje zmianę jej polaryzacji. Niektóre informacje otrzymane przez komórkę nerwową są hamujące — stłumiają aktywność komórki odbiorczej, inne z kolei działają pobudzająco - zwiększają jej czynność. W przypadku neurotransmiterów pobudzających jest to zmiana dodatnia powodująca depolaryzacje w błonie postsynaptycznej i wytworzenie pobudzającego potencjału postsynaptycznego. Neuroprzekaźniki hamujące - jest to zmiana ujemna, powodują hyperpolaryzację (komórka nie jest zdolna do przewodzenia impulsu i przekazywania informacji), powstawanie hamującego potencjału postsynaptycznego. Gdy neuroprzekaźnik spełni swoją funkcje zostaje unieczynniony przez specjalne dezaktywujące mechanizmy [3].
Najbardziej rozpowszechnionymi neuroprzekaźnikami są: acetylocholina, aminy biogenne (adrenalina, dopamina, noradrenalina, histamina, serotonina), aminokwasy (asparaginian, glicyna, glutaminian, tauryna, kwas γ-aminomasłowy), a także neuropeptydy (m.in. enkefaliny, endorfiny). Większa grupa tych substancji to neurotransmitery pobudzające. Głównymi transmiterami hamującymi są kwas γ-aminomasłowy (GABA) oraz glicyna. Pośrednio hamująco oddziałują peptydy opioidowe (enkefaliny) posiadające działanie przeciwbólowe [3].
Charakterystyka kwasu γ-aminomasłowego (GABA)
Kwas γ-aminomasłowy (GABA) (Rys.1) pełni funkcję głównego neuroprzekaźnika o działaniu hamującym w ośrodkowym układzie nerwowym. GABA w układzie nerwowym ssaków, w tym ludzi działa przeciwdrgawkowo, uspakajająco i nasenne, przeciwstresowo, przeciwlękowo oraz redukuje łaknienie. Oddziaływuje on także na system sercowo-naczyniowy oraz wpływa na wydzielanie hormonów (hamuje wydzielanie prolaktyny). Produkowanie GABA następuje przez neurony gabaergiczne w móżdżku i korze mózgu poprzez dekarboksylację glutaminianu, w której udział bierze enzym dekarboksylazy kwasu glutaminowego (GAD). Efektem tego procesu jest powstający GABA, gromadzony w pęcherzykach synaptycznych neuronów gabaergicznych. Tam też dzięki impulsowi uwalniany jest do przestrzeni synaptycznej, gdzie wywołuje swoje działanie hamujące poprzez reakcję ze specyficznymi receptorami błony postsynaptycznej synapsy. Do rozpadu GABA dochodzi w wyniku transaminacji do semialdehydu bursztynowego, który zostaje utleniony do kwasu bursztynowego i wchodzi w cykl Krebsa. Przemiana ta prowadzona jest przez enzym γ-aminotransferazy (GABA-T). Receptory GABA dzielą się na trzy rodzaje: GABAA, GABAC – są to receptory jonotropowe oraz GABAB – receptory metabotropowe. Receptor GABAA tworzy kanał chlorkowy, który jest zbudowany z pięciu podjednostek białkowych. Aktywność tego receptora regulowana jest wiązaniem swoistego liganda – kwasu γ- aminomasłowego. Związek ten prowadzi do aktywacji receptora i otwarcia kanału jonowego, co skutkuje zwiększeniem napływu jonów chlorkowych do wnętra komórki. Ze względu na to, że receptory te są umiejscowione w błonie komórkowej neuronów, napływ jonów CI- przyczynia się do wzrostu różnicy potencjałów po obu stronach błony, a więc jej hyperpolaryzację. Skutkiem tego jest utrudnienie powstawania czynnościowych potencjałów, które odpowiadają za przekazywanie informacji w układzie nerwowym. Z tego powodu GABA jest nazywany aminokwasem hamującym, a jego receptor – receptorem hamującym. Natomiast receptor GABAB jest powiązany poprzez białko G z kanałem potasowym, wapniowym, a także z cyklazą adenylanową i fosfolipazą C. Gdy są one zlokalizowane na presynaptycznych zakończeniach nerwowych, spełniają funkcję zarówno autoreceptorów jak i heteroreceptorów, regulując w ten sposób uwalnianie neuroprzekaźników. Agonistą tych receptorów jest baklofen - lek, który zwiotcza mięśnie szkieletowe. Wynikiem pobudzenia receptora GABAB jest powolne i przedłużone hamowanie uwalniania neuroprzekaźników [4].
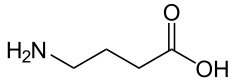
Rys. 1. Wzór strukturalny kwasu γ-aminomasłowego (GABA).
Z wielu badań wynika, że w chorobach degeneracyjnych ośrodkowego układu nerwowego, takich jak: schizofrenia, epilepsja, stany lękowe, choroba Parkinsona, Alzheimera oraz zaburzenia maniakalno-depresyjne, występuje zaburzenie funkcjonowania układu gabaergicznego. Zaburzenia te pojawiają się głównie przez obniżenie poziomu GABA w mózgu. Ze względu na charakterystyczną strukturę cząsteczki GABA, nie jest możliwe zaaplikowanie syntetycznego GABA u chorych. W warunkach doświadczalnych wykazano, że GABA działa szybko i odwracalnie z zastosowaniem miejscowym na powierzchnię kory mózgowej. Przy podaniu dożylnym, nawet w dość dużych stężeniach, nie dostrzega się identycznego wpływu elektrofizjologicznego, ponieważ bariera krew-mózg nie pozwala mu dotrzeć do miejsca docelowego, czyli mózgu [5].
GABA nie przekracza bariery krew-mózg ponieważ jest bardzo niestałą cząsteczką o wysokiej polarności. Bariera krew-mózg pełni funkcję ochronną dla mózgu przed szkodliwymi substancjami. Posiada ona znaczenie selektywne pomiędzy naczyniami krwionośnymi, a tkanką nerwową. Odpowiednią barierę krew-mózg tworzą wyspecjalizowane komórki pokrywające wewnętrzną powierzchnię naczyń włosowatych mózgu, które są ułożone tak ściśle, że nie dają możliwości swobodnego transportu substancji z krwi do mózgu. Różnią się więc od obwodowych naczyń krwionośnych, które posiadają duże przestrzenie między komórkami przez które mogą swobodnie przemieszczać się płyny do tkanek. Transport do mózgu substancji odżywczych, tlenu, dwutlenku węgla jest umożliwiony przez barierę krew-mózg. Stanowi ona jednak istotną przeszkodę w przenikaniu innych substancji (niektórych leków, hormonów), a w związku z tym leczenia chorób układu nerwowego [5].
Problem zaburzenia poziomu GABA w mózgu można rozwiązać przez zaprojektowanie analogu GABA. Trzeba jednak wziąć pod uwagę zmniejszenie jego niestałości, a także hydrofilowości. Można to osiągnąć poprzez wprowadzenie w jego strukturę usztywniających ugrupowań, przede wszystkim układów pierścieniowych. Cząsteczka GABA będzie wówczas posiadać większą skłonność do przekroczenia bariery krew-mózg [5].
Analogi GABA w zwalczaniu epilepsji
Epilepsja to najczęściej spotykana choroba układu nerwowego, którą cechują nawracające napady drgawkowe, pojawiające się w skutek nagłych, krótkotrwałych, przewlekłych zaburzeń czynności mózgu (w okolicy kory mózgowej i części podkorowej). Zaburzenia te wynikają z nadmiernych i gwałtownych wyładowań elektrycznych w komórkach nerwowych. Uszkodzenie mózgu prowadzące do epilepsji może nastąpić w każdym wieku. Takimi przykładami mogą być: uraz okołoporodowy, wady wrodzone, niedotlenienie mózgu, zmiany naczyniowe lub ropne, zapalenie mózgu i opon mózgowych, urazy głowy oraz nowotwory mózgu. W przypadku około 70% chorych pierwsze objawy choroby pojawiły się w dzieciństwie. Napad padaczkowy może być sprowokowany za pomocą różnych czynników na przykład elektrycznych bądź chemicznych, natomiast wyładowanie padaczkowe może wystąpić w rozmaitych populacjach komórek nerwowych mózgu. W takich ogniskach padaczkowych komórki dostają zbyt dużą ilość pobudzających bodźców. Skutkiem tego jest nagła depolaryzacja błony komórkowej, która może tworzyć się przez patologiczne zmiany morfologiczne. Można do nich zaliczyć: zaburzenia błonowej regulacji przepływu jonów potasowych, sodowych, wapniowych, a także układu neuroprzekaźników, a głównie GABA. Wyróżniamy kilkadziesiąt typów epilepsji, a dla około jednej trzeciej brak jest skutecznych leków [6].
Wieloletnie badania nad neurochemicznymi mechanizmami napadów padaczkowych dowodzą udział w tych procesach czynników zewnątrzkomórkowych (wydzielaniu neuroprzekaźników i neuromodelatorów, zmiany w biosyntezie), jak również czynników błonowych (zaburzenia w czynnościach pomp, kanałów jonowych i receptorów), a także czynników wewnątrzkomórkowych (zaburzenia homeostazy wapnia, metabolizmu oraz stanu energetycznego komórki). Naukowcy potwierdzili teorię, że do ataków padaczki dochodzi w wyniku obniżenia się w mózgu poziomu GABA [7].
Ze względu na wiele przyczyn powstawania padaczki, jest małe prawdopodobieństwo wynalezienia jednego leku na tę chorobę. Całkowite zapobieganie powstawaniu epilepsji jest również nie możliwe, gdyż pewna grupa przypadków jest dziedziczna, wobec czego są małe szanse na usunięcie nieprawidłowych genów za pomocą technik inżynierii genetycznej. Około 25-30% chorych należy do grupy opornych na leczenie, to znaczy wymagają stosowania więcej niż jednego leku. Jest to powód, dla którego wciąż prowadzi się badania w poszukiwaniu większej skuteczności podstawowych leków, pozwalających zredukować grupę pacjentów z oporną na leki postacią padaczki. Nowa generacja leków, głównie analogi GABA, są zaprojektowane do leczenia epilepsji [7].
Leki przeciwpadaczkowe mogą działać na dwa sposoby: leki, które stabilizują błony komórkowe neuronów przeciwdziałając rozprzestrzenianiu się impulsów, bądź leki przywracające równowagę pomiędzy ilością neuroprzekaźników pobudzających i hamujących funkcje neuronów. Pojawienie się w ostatnich latach na rynku farmaceutycznym nowych leków stwarza dla neurologów zajmujących się epilepsją doskonałe pole do działania terapeutycznego [7].
Najnowszym osiągnięciem w dziedzinie badań nad analogami GABA było odkrycie, że Baklofen (Rys.2) jest agonistą receptora GABAB, który wywiera hamujący wpływ w ośrodkowym układzie nerwowym. Baklofen (Kemstro, baclo-fenum, Lioresal, kwas (R,S)-β-(4-chlorofenylo)-γ-aminomasłowy) jest pochodną kwasu γ-aminomasłowego. Po raz pierwszy został zsyntetyzowany w 1962 roku. Zaprojektowano go jako lek przeciwko padaczce. W porównaniu z GABA podstawnik 4-chlorofenylowy wpływa na dwukrotne zwiększenie lipofilowości tego związku, dlatego pomimo struktury aminokwasowej ma możliwość przenikania do ośrodkowego układu nerwowego. Baklofen stosuje się głównie w celu zmniejszenia napięcia mięśniowego w stwardnieniu rozsianym, chorobach rdzenia kręgowego oraz po urazach [8].
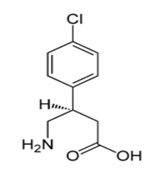
Rys.2. Wzór strukturalny Baklofenu.
Wigabatryna (Rys.3) (Vigabatrinum, Sabril, Wigabatrina, kwas γ-winyloaminomasłowy) jest strukturalnym analogiem GABA. Lek ten zwiększa stężenie GABA w przestrzeni synaptycznej hamując aktywność transaminazy GABA (GABA-T), enzymu rozkładającego neuroprzekaźnik GABA. Zwiększenie w mózgu stężenia GABA nawet o 30%, powoduje jej działanie przeciwdrgawkowe. Wigabatryna jest racemidem posiadającym dwie formy enancjomeryczne. Tylko forma (+) - S jest aktywna farmakologicznie. Wigabatryna jest jednym z najskuteczniejszych leków przeciwpadaczkowych, jakie zostały w ostatnich latach wprowadzone na rynek farmakologiczny [8,9].
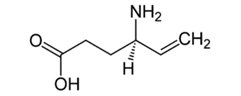
Rys.3. Wzór strukturalny Wigabatryny. Gabapentyna (Rys.4) (Gabapentinum, Neurontin, kwas 1-(amino-metylo)-cykloheksano-1-octowy) uważana jest za ośrodkowo działający analog GABA, który wskutek obecności lipofilnego pierścienia cykloheksanowego z łatwością przenika przez barierę krew-mózg. Gabapentyna ma wpływ na syntezę i uwalnianie GABA w strukturach mózgowych. W ośrodkowym układzie nerwowym Gabapentyna wiąże się z receptorem białkowym (białkiem wiążącym gabapentyne) i przez skomplikowany i nie do końca poznany mechanizm zwiększa produkcję i uwalnianie GABA. Efektem tego jest nasilenie przewodnictwa GABA-ergicznego, co w następstwie powoduje zahamowanie aktywności bioelektrycznej neuronów. Mechanizm działania Gabapentyny jest więc inny niż leków wpływających na przekaźnictwo gabaergiczne [8,10].
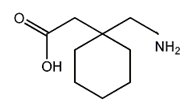
Rys.4. Wzór strukturalny Gabapentyny.
Pregabalinę (Rys.5) (Pregabalinum, Lyrica, (S)-3-izobutylo-GABA) zaprojektowano jako lipofilowy analog GABA podstawiony w trzeciej pozycji w celu ułatwienia dyfuzji GABA poprzez barierę krew-mózg. Miało to wpłynąć na wzrost aktywności dekarboksylazy kwasu glutaminowego (enzym, który odpowiada za biosyntezę GABA), a jednocześnie podniesienie poziom GABA w mózgu. W dalszych badaniach prowadzonych przez firmę farmaceutyczną Pfizer, która kupiła licencję na korzystanie ze związku wykazano, że Pregabalina posiada wysoką aktywność przeciwpadaczkową. Odkryto, że działanie Pregabaliny polega na łączeniu się leku z podjednostką jednego z rodzajów kanałów wapniowych, jakie występują w błonach komórek nerwowych. Kanał wapniowy sprawdza uwalnianie kwasu glutaminowego z zakończeń nerwowych, powodując wzrost stężenia GABA w komórkach nerwowych [8,10].
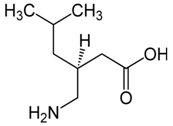
Rys.5. Wzór strukturalny Pregabaliny.
Tiagabina (Rys.6) (tiagabinum, kwas (3R)-1-[4,4-bis(3-metylo-2-tiofeno)-3-butenylo]-3-piperydynokarboksylowy) mieści w swej cząsteczce kwas nipekotynowy (3-piperydynokarboksylowy), który połączony jest mostkiem butanowym z dwoma lipofilowymi pierścieniami 3-metylotiazolu. Tiagabina jest specyficznym inhibitorem wychwytu kwasu γ-aminomasłowego w komórkach glejowych oraz zakończeniach nerwowych. Wzrost stężenia GABA w płynie zewnątrzkomórkowym oraz nasilenie neurotransmisji zależnej od GABA prowadzi do zahamowania wychwytu. Reasumując działanie Tiagabiny polega na hamowaniu wychwytu zwrotnego GABA do zakończeń nerwowych i gleju, zatem zwiększając stężenie GABA w synapsie. Lek ten daje dobre rezultaty w zapobieganiu wielu typom napadów drgawkowych w modelach zwierzęcych [8,10].
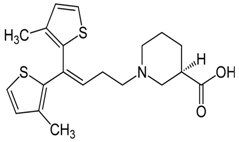
Rys.6. Wzór strukturalny Tiagabiny.
Progabid (Rys.7) (progabidum, Gabrene, 4-{[4-chlorofenylo)-5-fluoro-2-hydroksyfenylo)-metyleno]-amino}-butanamid), jest pierwszym selektywnym agonistą receptorów GABA-ergicznych. Lek ten jest zasadą Schiffa (służy jako gabamimetyk) utworzoną na skutek reakcji od-powiednio podstawionego benzofenonu i γ-aminobutyroamidu. Wprowadzenie cząsteczki benzofenonu, spowodowało zwiększenie właściwości lipofilowych, utrzymując w równowadze hydrofilowy charakter GABA. Przez zmianę końcowej grupy aminowej GABA na wiązaniu iminowym nastąpiło zwiększenie trwałości hydrolitycznej cząsteczki oraz zmniejszenie zasadowości, przez co jest ona mniej polarna. Progabid będąc agonistą receptorów GABA–ergicznych, stymuluje te receptory doprowadzając do zwiększonego napływu jonów chlorkowych do komórki i jej hyperpolaryzację, co prowadzi do zahamowania przewodzenia impulsu [8,11].
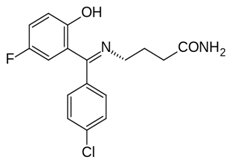
Rys.7. Wzór strukturalny Progabidu.
Podsumowanie:
Niski poziom GABA w chorobach układu nerwowego, głównie w epilepsji, depresji, schizofrenii, lękach, chorobie Parkinsona czy Alzheimera oraz brak możliwości transportowania GABA przez barierę krew-mózg, przyczyniło się do poszukiwania i projektowania analogów opartych na strukturze GABA.
W ostatnich kilkunastu latach nastąpił duży postęp w epileptologii, obejmujący wiele dziedzin nauki takich jak: neurologia, neurofarmakologia, neurofizjologia, psychologia, neurodiagnostyka czy też neurochirurgia. Wynikiem tego jest zwiększenie jakości opieki zdrowotnej większej grupy chorych cierpiących na epilepsje. Głównym celem leczenia tej choroby jest niesienie pomocy pacjentowi w dostosowaniu się do wymogów życia. Naukowcy w prowadzonych badaniach skupiają się na minimalizacji liczby napadów, zapobieganiu ich nawrotowi, zmniejszeniu ciężkości napadów, a także poprawę funkcjonowania psychospołecznego chorych na epilepsję. Obecnie powiększa się dostępność nowych leków przeciwpadaczkowych oraz poprawia prawidłowość w postępowaniu terapeutycznym. Zastosowanie kilku rodzajów padaczki pomogło w określeniu profilu farmaceutycznego nowych leków oraz przewidywania działań na różne typy epilepsji. Ma to duże znaczenie w terapii przeciwpadaczkowej w leczeniu poszczególnych rodzajów napadów. Pomimo, że leki nowej generacji stosowane są dość krótko, na podstawie badań klinicznych stwierdzono przydatność niektórych substancji leczniczych w pewnych typach padaczek i w leczeniu napadów padaczkowych szczególnie u osób z zaburzeniami psychicznymi. Stosowane na rynku farmaceutycznym leki przeciwpadaczkowe wywołują także poważne działania niepożądane, w dużym stopniu utrudniając życie chorym. Poza tym nawet przy stosowaniu znanych antyepileptyków nadal jeszcze u około 20%, a być może znacznie większego odsetka pacjentów, skuteczne hamowanie napadów padaczkowych jest niemożliwe. Jest to powód, dla którego wciąż projektuje się wiele nowych analogów GABA, dołączając do cząsteczki GABA element transportujący poprzez barierę krew-mózg w postaci ugrupowania lipofilnego. Stosowane obecnie w farmakologii analogi są często modyfikowane, korzysta się również z produktów pochodzenia naturalnego mając nadzieję na brak toksyczności otrzymanych tym sposobem analogów GABA.
Autor: Katarzyna Czuba
Literatura:
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje