
|

Zamknij X
|




W ostatnich latach pojawiła się nowa hipoteza, która mówi, że nowotwór wywodzi się ze stransformowanych komórekposiadających cechy komórek macierzystych. Zgodnie z ta hipotezą nowotwór zawiera niewielką liczbę niezróżnicowanych lub słabo zróżnicowanych komórek macierzystych nowotworu (KMN). Komórki te są zdolne do ciągłej proliferacji oraz samoodnowy w wyniku podziału niesymetrycznego. Z tych komórek powstają następnie bardziej zróżnicowane komórki nowotworowe, stanowiące większość populacji nowotworu. Hipoteza ta wyjaśnia, dlaczego nowotwór składa się z komórek różniących się cechami morfologicznymi, biochemicznymi oraz posiadających różną wrażliwość na czynniki terapeutyczne. Hipoteza ta tłumaczy także przyczyny niepowodzeń w leczeniu nowotworów.
Podczas standardowej terapii przeciwnowotworowej początkowo następuje spadek ilości komórek nowotworowych u pacjenta, często do poziomu niewykrywalnego nowoczesnymi metodami diagnostycznymi. Jednak po pewnym czasie obserwuje się odbudowę pierwotnego ogniska nowotworu, ponieważ nie udało się wyeliminować komórek macierzystych nowotworu, opornych na stosowane terapie. Prawdopodobnie komórki KMN odpowiadają również za tworzenie przerzutów poprzez zwiększoną zdolność do migracji i adaptacji do nowych warunków środowiska w nowej tkance [1].
Pochodzenie komórek macierzystych nowotworu (KMN)
Istnieje kilka hipotez na temat pochodzenia komórek macierzystych nowotworu (KMN). Według pierwszej hipotezy komórki KMN powstają w wyniku zmian w pluripotentnych komórkach macierzystych, bądź częściowo już zróżnicowanych komórkach progenitorowych (tkankowo-specyficznych komórkach macierzystych). Komórki te mogą ulec transformacji nowotworowej, przy jednoczesnym zachowaniu zdolności do samoodnowy i niesymetrycznych podziałów, podczas których powstają bardziej zróżnicowane komórki nowotworowe oraz odtwarzane są komórki macierzyste nowotworu. Kolejna hipoteza mówi, że powstanie stransformowanych komórek macierzystych może być powodowane częściowym odróżnicowaniem komórek somatycznych, które pod wpływem różnych czynników uległy zmianom genetycznym i epigenetycznym. Na skutek tych procesów komórki te mogą nabyć cech komórek macierzystych, które są zdolne do niekontrolowanego wzrostu, co następnie prowadzi do powstania nowotworu [2].
Oporność komórek macierzystych nowotworu (KMN) na terapie przeciwnowotworowe
Komórki macierzyste nowotworu są bardziej odporne na chemoterapię oraz radioterapię. Prawdopodobnie przyczyna tkwi w niepowodzeniu standardowych terapii i skutkuje nawrotem nowotworu. Typowa terapia eliminuje jedynie proliferujące komórki (zróżnicowane komórki nowotworowe), natomiast „uśpione" KMN są oporne na chemoterapeutyki i po zakończeniu terapii zaczynają proliferować oraz różnicować się, powodując remisję nowotworu. Terapia eliminująca KMN powoduje zabicie zarówno proliferujących, jak i „uśpionych" KMN, co skutkuje zmniejszaniem ilości komórek nowotworowych oraz daje możliwość ich eliminacji standardowymi chemoterapeutykami [3].
Oporność komórek macierzystych nowotworu na standardowe terapie jest związana z opornością wielolekową tych komórek oraz nieuleganiu apoptozie. Zjawiska te są spowodowane wieloma zmianami genetycznymi i adaptacyjnymi KMN, takich jak zwiększona wydajność systemów naprawy uszkodzeń DNA, zwiększona ekspresja transporterów błonowych oraz spowolnienie kinetyki progresji cyklu komórkowego (Rys. 1) [3].
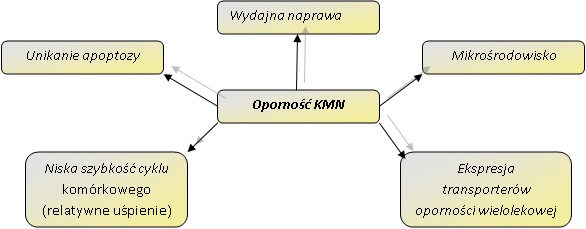
Rys. 1. Przyczyny oporności komórek macierzystych nowotworu na chemoterapię i radioterapię.
Komórki macierzyste nowotworu szybciej odpowiadają na uszkodzenia DNA, wskutek zwiększonej aktywności kinaz punktów kontrolnych cyklu komórkowego Chk1 i Chk2 oraz kinazy ATM. Zastosowanie inhibitorów kinaz Chk 1 oraz Chk2 doprowadziło do zwiększenia wrażliwości komórek glejaka na radioterapię [4].
Usuwanie chemoterapeutyków z komórek macierzystych nowotworu jest związane z zwiększoną ekspresją transporterów błonowych typu ABC, takich jak ABCG2 (ang. Breast Cancer Resistance Protein - BCRP) i ABCB1 (ang. Multidrug Resistance Protein 1 – MDR1). Badania subpopulacji komórek białaczek wzbogaconej w KMN, wykazały zwiększoną zdolność do wypompowywania z wnętrza komórki chemoterapeutyków, takich jak daunorubicyna i mitoksantron. Zwiększona zdolność do usuwania leków może prowadzić do wielolekooporności komórek macierzystych nowotworu [5].
Komórki macierzyste nowotworu nadprodukują także molekularne mediatory metaboliczne, takie jak dehydrogenaza aldehydowa 1 (ALDH1). Związek ten jest odpowiedzialny za oporność zarówno normalnych komórek macierzystych jaki i KMN na cyklofosfamid w białaczkach. Wynika z tego, że mechanizm chemiooporności obecny w KMN bezpośrednio wpływa na skuteczność terapii [6].
Wrażliwość komórek nowotworowych na chemoterapeutyki uszkadzające DNA zależy również od tempa ich proliferacji, które z kolei jest związane z kinetyką progresji w cyklu komórkowym. Komórki szybko proliferujące są bardziej narażone na letalne uszkodzenia DNA, powodowane działaniem chemoterapeutyków, niż wolno proliferujące komórki macierzyste. Przykładowo KMN ostrej i przewlekłej białaczki mielocytarnej pozostają uśpione, co przyczynia się do ich oporności na terapię. Prawdopodobnie najmniej zróżnicowane komórki nowotworowe „naśladują" normalne komórki macierzyste i tak jak one odznaczają się niskim tempem proliferacji i samoodnowy, przyczyniając się tym samym do ich oporności na chemoterapeutyki [7].
Molekularne cele dla eliminacji komórek macierzystych nowotworu (KMN)
Standardowe terapie przeciwnowotworowe eliminują tylko szybko proliferujące i stosunkowo wrażliwe na czynniki terapeutyczne zróżnicowane komórki nowotworowe. Natomiast nie eliminują komórek macierzystych nowotworu. Jedną z możliwych strategii terapeutycznych ukierunkowaną na usuwanie KMN jest analiza przyczyn oporności i charakteryzacja mechanizmów oporności KMN na stosowane terapeutyki. Inna metoda polega na przeszukiwaniu bibliotek wybranych związków, o różnych mechanizmach działania, pod kątem znalezienia cząsteczki, która posiadałaby selektywną toksyczność wobec komórek macierzystych nowotworu. Jednakże znalezienie takich nowych leków może być bardzo trudne [8].
Celem molekularnym dla terapii przeciwnowotworowych mogą być szlaki sygnałowe odpowiedzialne za zdolność komórek macierzystych do ich samoodnowy i różnicowania. Wykazano, że nieprawidłowe działanie szlaków sygnałowych lub ich nadmierna aktywacja mogą być przyczyną nowotworzenia. Szlaki sygnałowe Wnt, Notch i Hedgehog odgrywają istotną rolę w utrzymaniu właściwości zarówno normalnych jak i nowotworowych komórek macierzystych. Wykrycie różnic w powyższych szlakach sygnałowych pomiędzy normalnymi a nowotworowymi komórkami macierzystymi umożliwiłoby stworzenie selektywnej terapii skierowanej przeciwko KMN przy jednoczesnym uniknięciu efektów ubocznych powodowanych zahamowaniem funkcji normalnych komórek macierzystych [8].
Szlak Wnt należy do głównych szlaków sygnałowych występujących w nowotworach. Odgrywa on istotną rolę w regulacji takich procesów jak embriogeneza, różnicowanie, proliferacja i przeżywalność komórek. Przekazywanie sygnału za pomocą ścieżek Wnt zależy od rodzaju liganda Wnt oraz warunków panujących w komórce. Wyróżniamy dwie ścieżki sygnałowe: kanoniczną (zależną od β-kateniny) oraz niekanoniczną (zależną m.in. od stężenia jonów wapnia). Kanoniczna ścieżka sygnałowa Wnt wpływa na proces nowotworzenia i proliferację chorobowych komórek. Przyczyny aktywacji szlaku Wnt są wynikiem mutacji poszczególnych białek tej ścieżki lub wyciszania ekspresji negatywnych regulatorów Wnt/β-kateniny takich jak DKK lub WIF1. Do aktywacji szlaku sygnałowego Wnt/β-katenina dochodzi wówczas gdy białko Wnt zwiąże się z koreceptorem LRP5/6 i aktywowanym receptorem Frizzled (Rys.2). Fosforylacja β-kateniny zostaje zahamowana, co zapobiega jej degradacji. Następnie β-katenina przechodzi z cytoplazmy do jądra gdzie tworzy kompleks z TCF/LEF (ang. transcription factor/lymphoid enhancer binding factor), PYG (ang. Pygopus), BCL9 (ang. B-cell CLL/lymphoma 9 protein), CBP (ang. cAMP-response element-binding protein (CREB)-binding protein) oraz p300, aktywując transkrypcję genów docelowych, takich jak cyklina D1, C-Jun, c-Myc czy fibronektyny. Kaskada Wnt stanowi istotny regulator wzrostu komórek macierzystych i samoodnowy. Deregulacja szlaku sygnalizacyjnego Wnt/β-katenina prowadzi do powstania proliferujących, zdolnych do migracji i tworzenia przerzutów komórek nowotworowych. Badania przeprowadzone na myszach wykazały, że zaburzenia Wnt1 mogą prowadzić do powstania nowotworów piersi, a nadmierna ekspresja tego genu przyczynia się do odporności komórek na działanie leków. Natomiast nadekspresja Wnt5 powoduje uzłośliwienie nowotworów, zwiększając ruchliwość i proliferację zmienionych komórek. Obecnie do badań przedklinicznych zostały zakwalifikowane nowe inhibitory szlaku Wnt/β-katenina, takie jak PRI-724 blokujący interakcje pomiędzy β-kateniną a CBP, CWP232291 promujący degradację β-kateniny oraz XAV939 deregulujący szlak Wnt/β-kateniny [8].
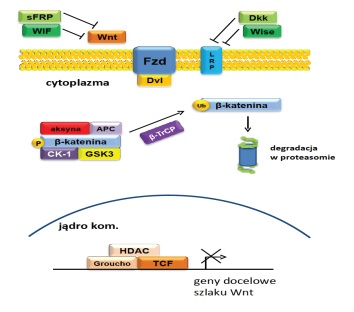
Rys.2. Aktywny szlak sygnałowy Wnt.
Szlak sygnałowy Notch ma istotne znaczenie dla rozwoju i utrzymania homeostazy tkanek. Odpowiada on za regulację procesów związanych z nabywaniem określonego fenotypu podczas różnicowania komórek oraz kontrolę przeżywalności i interakcji międzykomórkowych. Ścieżka sygnałowa Notch zostaje aktywowana, gdy ligand DSL (Delta, Serrate, Lag2) zwiąże się z białkiem Notch, indukując serię cięć - S2, S3 i S4 (Rys.3). Cięcie S2 dokonywane jest przez metaloproteazę ADAM, natomiast wewnątrzmembranowe cięcia S3 i S4 dokonywane są przez kompleks γ-sekretazy. Skutkuje to przemieszczeniem wewnątrzkomórkowej domeny Notch (NICD) do jądra, gdzie następnie wiąże się ona z czynnikiem transkrypcyjnym CSL, aktywując transkrypcję genów docelowych, takich jak rodzina genów Hes. Produkty tych genów są odpowiedzialne za regulację wzrostu i różnicowania komórek. Ścieżka Notch dostarcza sygnału proliferacyjnego komórka-komórka w embrionalnych komórkach macierzystych (ang. embryoic stem cells - ESC) oraz w ich złośliwych odpowiednikach (ang. embryonal carcinoma - EC) oraz odgrywa istotną rolę w utrzymaniu niezróżnicowanych komórek ESC i EC. Zaburzenia poziomu białka Notch1 i Notch2 udokumentowano w nowotworze szyjki macicy, jelita grubego, trzustki, skóry oraz mózgu. Natomiast wzmożona synteza Notch3 i Notch4 występuje w nowotworze trzustki i w czerniaku. Obecnie w badaniach klinicznych testowane są małocząsteczkowe inhibitory γ-sekretazy, takie jak MK0752 i R04929097. Ponadto w I fazie badań klinicznych znajduje się przeciwciało monoklonalne OMP-21M18, jako potencjalny lek do leczenia raka trzustki [9].
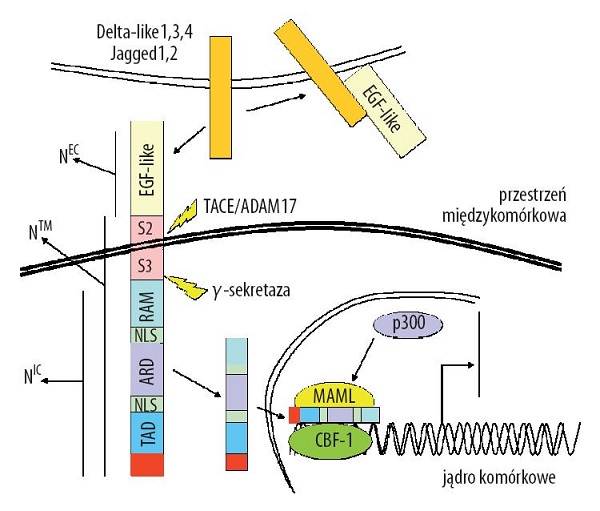
Rys. 3. Szlak sygnałowy Notch.
Szlak sygnałowy Hedgehog odgrywa ważną rolę w embriogenezie i regulacji komórek macierzystych, natomiast w sytuacjach patologicznych indukuje proces nowotworowy. Nadekspresja białka Shh na błonie komórek nowotworowych pobudza je autokrynnie do stałej proliferacji, powodując progresję nowotworu. Szlak Hedgehog składa się z trzech głównych komponentów: cytoplazmatycznego kompleksu regulującego czynniki transkrypcyjne z rodziny Gli, liganda Hh oraz transmembranowych receptorów: receptora Ptc działającego jako negatywny regulator oraz aktywatora Smo. Aktywacja ścieżki sonic hedgehog następuje w wyniku połączenia aktywnego białka Shh z receptorem Ptch i aktywacji SMO, co powoduje rozszczepienie białek Gli aktywujących geny docelowe (Rys. 4). W prawidłowych komórkach macierzystych aktywacja białka SMO zwiększa transkrypcję Ptch1 i prowadzi do zahamowania procesu proliferacji. Mutacja jednego ze składowych szlaku lub przedostanie się w całości do jądra białka Gli, może spowodować ciągłą proliferację komórkową i inicjować tworzenie nowotworów. Szlak Hedgehog odgrywa istotną rolę w kontroli proliferacji i różnicowaniu zarówno ESC, jak i somatycznych komórek multipotencjalnych. Dlatego też zmiany, zakłócające system sygnalizacji Hh, mogą powodować rozwój wielu rodzajów nowotworów. Jednym z inhibitorów szlaku Hedgehog, który posiada właściwości leków i charakteryzuje się wysoką aktywnością jest cyklopamina. Preparat ten może być potencjalnym chemoterapeutykiem do leczenia nowotworów, takich jak rak podstawno-komórkowy i rdzenia. Ponadto wyodrębniono dwa potencjale leki będące antagonistami białka Smo: SAG, Cur-61414 i GDC-0449 [10].
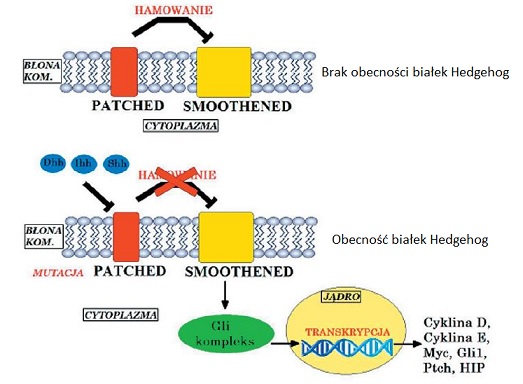
Rys. 4. Szlak Hedgehog.
Drugim podejściem przy poszukiwaniu nowych związków o selektywnym działaniu anty-KMN są badania przesiewowe przy wykorzystaniu dużych bibliotek cząsteczek o różnych mechanizmach cytotoksycznych. Dotychczas wyselekcjonowano kilka związków, które zwiększają skuteczność terapii przeciwnowotworowej poprzez wpływ na KMN. Należą do nich m.in. związki chemiczne blokujące metabolizm komórkowy. Jednym z takich związków jest salinomycyna (jonofor jonu potasowego), która wykazała ponad 100-krotnie wyższą cytotoksyczność wobec komórek KMN raka sutka w porównaniu ze standardowymi lekami, takimi jak paklitaksel [11]. Kolejnym związkiem, który może być zastosowany w kombinacji z powszechnie stosowanymi lekami jest metformina, używana w leczeniu cukrzycy typu drugiego. Podstawowy mechanizm działania metforminy polega na zmniejszeniu produkcji glukozy w organizmie poprzez hamowanie szlaku kinaz AMPK/mammalian target of rapamycin (AMPK/mTOR). Stymuluje ona także proces glikolizy poprzez działanie na transportery GLUT 1/4. Badania przedkliniczne wykazały, że metformina posiada aktywność przeciwnowotworową wobec wielu typów nowotworów, takich jak rak sutka, płuc, wątroby i trzustki. Ponadto badania prowadzone na modelach komórek nowotworowych w hodowli in vitro i na modelach zwierzęcych wykazały, że metformina jest zdolna do uwrażliwiania KMN na działanie różnych czynników terapeutycznych, takich jak doksorubicyna, trastuzumab oraz promieniowanie jonizujące [12]. Inną cząsteczką znalezioną w badaniach przesiewowych i wykazującą toksyczność wobec KMN jest tiorydazyna, lek neuroleptyczny z grupy piperydynowych pochodnych fenotiazyny. Wykazano, że tiorydazyna selektywnie hamuje wzrost KMN w białaczkach i komórkach raka sutka poprzez blokowanie receptorów dopaminowych znajdujących się na powierzchni błony komórek KMN, co w konsekwencji prowadzi do ich różnicowania i powstrzymuje ich wzrost [13].
Istnieją także doniesienia, że komórki macierzyste nowotworu (KMN) posiadają zmienione szlaki regulacji punktów kontrolnych cyklu komórkowego, szczególnie te aktywowane w odpowiedzi na uszkodzenia DNA podczas przejścia metafaza-anafaza oraz w fazie S i G2. Istnieje zatem możliwość terapii skojarzonymi modulatorami aktywności punktów kontrolnych i czynnikami uszkadzającymi DNA. Przykładowo zwiększona aktywność kinaz Chk1 i Chk2 w komórkach KMN glejaka doprowadziła do zwiększonej aktywacji punktu kontrolnego w fazie G2 i ich radiooporności. Zahamowanie aktywności tych kinaz za pomocą chemicznych inhibitorów, takich jak debromohymenialdisine czy AZD7762, zwiększyło wrażliwość KMN glejaka na działanie gemcitabiny oraz promieniowania jonizującego [14].
Oporność komórek macierzystych nowotworu (KMN) na chemo- i radioterapię może również wynikać z aktywności szlaku sygnalizacyjnego blokującego apoptozę związanego z kinazami PI3K/Akt oraz szlakiem zależnym od kinaz ATR/ATR, uruchamianym w obecności uszkodzeń DNA. Prawdopodobnie regulacja odpowiedzi komórkowej na uszkodzenia DNA w KMN jest podobna do tej uruchamianej w komórkach normalnych i zależy od aktywności kinaz ATR/ATM. Dotychczas potwierdzono rolę szlaku kinazy Akt w KMN dla ich inwazyjności oraz w odpowiedzi na terapie przeciwnowotworowe dla nowotworów płuc, jelita grubego, raka piersi i glejaków. Istnieje zatem możliwość eliminacji KMN za pomocą selektywnych inhibitorów szlaku kinaz PI3K/Akt stosowanych w połączeniu z czynnikami uszkadzającymi DNA, takimi jak cisplatyna, etopozyd oraz promieniowanie jonizujące [15].
Podsumowanie
W przypadku niektórych typów nowotworów istnieje możliwość wieloletniego przeżycia pacjentów bez symptomów choroby w wyniku zastosowanej terapii. Przykład mogą stanowić pewne typy białaczek, szczególnie diagnozowanych w młodym wieku, lub rak jądra u mężczyzn. Z drugiej strony, istnieją również nowotwory, takie jak rak sutka, prostaty, płuc czy mózgu, wobec których standardowe leki są mało skuteczne lub nieskuteczne. Wiąże się to z opornością komórek nowotworowych na chemio- i radioterapię, wynikającą z funkcjonowania w komórkach nowotworowych mechanizmów oporności na czynniki terapeutyczne. Może to wynikać z istnienia dwóch różniących się między sobą grup nowotworów. W pierwszej grupie liczba komórek macierzystych nowotworu (KMN) jest niewielka lub komórki te wykazują podobną wrażliwość na terapię jak zróżnicowane komórki nowotworu. Natomiast druga grupa nowotworów posiada także komórki macierzyste nowotworu (KMN), ale oporne na działanie standardowych leków. W tym przypadku remisja nowotworu może wiązać się ze wzrostem liczby KMN po terapii oraz pojawieniem się dodatkowych zmian genetycznych lub epigenetycznych, co prowadzi do jeszcze większej oporności tych komórek na terapeutyki.
Badania nad komórkami macierzystymi mają istotne znaczenie w leczeniu chorób nowotworowych. Szczególnie niebezpieczne są nowotwory złośliwe ze skłonnością do tworzenia przerzutów do innych tkanek oraz zdolne do remisji po zastosowanym leczeniu. Z tego powodu, głównym celem terapii przeciwnowotworowej powinny być komórki macierzyste nowotworu, dla których standardowe terapie takie jak radioterapia czy chemioterapia są niewystarczająco skuteczne. Istotne znaczenie ma wyjaśnienie roli czynników regulujących szlaki proliferacji i różnicowania komórkowego, którego uszkodzenia mogą być przyczyną inicjacji i progresji nowotworów, ale również tworzenia przerzutów. Zahamowanie proliferacji tych komórek spowoduje zmniejszenie masy guza i zatrzymanie rozwoju choroby nowotworowej. Przeprowadzone badania wykazują, że zablokowanie szlaku sygnałowego Notch powoduje zredukowanie liczby komórek CSC, co w konsekwencji zatrzyma proces formowania guza. Ponadto zastosowanie cyklopaminy, która wiąże białko SMO szlaku Hedgehog wywołuje uwrażliwienie komórek macierzystych nowotworu na radioterapie. Udowodniono również, że nieprawidłowa sygnalizacja Wnt powoduje transformacje normalnych komórek macierzystych w nowotworowe i inicjuje proces powstawania nowotworu.
Dotychczas uzyskano obiecujące wyniki, które potwierdzają możliwość poprawy skuteczności chemio- i radioterapii poprzez zastosowanie podejść terapeutycznych celujących w komórki macierzyste nowotworu (KMN). Pomimo to, ciągle posiadamy zbyt mało informacji na temat KMN oraz zróżnicowanych komórek nowotworu, szczególnie jeśli chodzi o możliwość przechodzenia jednego fenotypu w drugi. Pewne jest, że skuteczna terapia przeciwnowotworowa musi zawierać leki, które w równym stopniu celują w komórki zróżnicowane, stanowiące większość populacji nowotworu, jak i eliminują komórki macierzyste nowotworu (KMN).
Literatura:
1.Wicha M.S., Liu S., Dontu G. Cancer stem cells: an old idea - a paradigm shift. Cancer Res. 2006. 66, 1883-1890.
2.Morrison S.J., Qian D., Jerebek L., Thiel B.A., Park I.K., Ford P.S. A genetic determinant that specifically regulates the frequency of hematopoetic stem cells. J. Immunol. 2002. 168, 635-642.
3.Dean M., Fojo T., Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer. 2005. 5, 275-284.
4.Eyler C.E., Rich J.N. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008. 26, 2839-2845.
5.Massard C., Teutsch E., Soria J.C. Tumour stem cell-targeted treatment: elimination or differentiation. Ann. Oncol. 2006. 17, 1620-1624.
6.Magni M., Shammah S., Schiro R. Induction of cyclophosphamide-resistance by aldehydedehydrogenase gene transfer. Blood. 1996. 87, 1097-1103.
7.Guan Y., Hogge D.E. Proliferative status of primitive hematopoietic progenitors from patients with acute myelogenous leukemia (AML). Leukemia. 2000. 14, 2135- 2141.
8.Pergoł P., Nowak-Stępniowska A., Drela K., Padzik-Graczyk A. Znaczenie komórek macierzystych w inicjacji i rozwoju nowotworów. Postępy biochemii. 2013. 59, 45-52
9.Radtke F., Raj K. The role of Notch in tumourigenesis: oncogene or tumour suppressor? Nat. Rev. Cancer. 2003. 3, 756-767.
10.Medina V., Calvo M.B., Diaz-Prado S., Espada J. Hedgehog signaling as target in cancer stem cells. Clin. Transl. Oncol. 2009. 11, 199-207
11.Gupta P., Onder T.T., Jiang G., Tao K., Kuperwasser C., Weinberg R.A., Lander E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009. 138, 645-659.
12.Ben Sahra I., Le Marchand-Brustel Y., Tanti J.F., Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug?Mol. Cancer Ther. 2010. 9, 1092- 1099.
13.Sachlos E., Risueno R.M., Laronde S., Shapovalova Z., Lee J.H., Russell J., Malig M., McNicol J.D., Fiebig-Comyn A., Graham M., Levadoux-Martin M., Lee J.B., Giacomelli A.O., Hassell J.A., Fischer-Russell D., Trus M.R., Foley R., Leber B., Xenocostas A., Brown E.D., Collins T.J., Bhatia M. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012, 149, 1284-1297.
14.Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D., Rich J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006. 444, 756-760.
15.Mueller M.T., Hermann P.C., Witthauer J., Rubio-Viqueira B., Leicht S.F., Huber S., Ellwart J.W., Mustafa M., Bartenstein P., D'Haese J.G., Schoenberg M.H., Berger F., Jauch K.W., Hidalgo M., Heeschen C. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009. 137, 1102-1113.
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje