



- Biochemia
- Biofizyka
- Biologia
- Biologia molekularna
- Biotechnologia
- Chemia
- Chemia analityczna
- Chemia nieorganiczna
- Chemia fizyczna
- Chemia organiczna
- Diagnostyka medyczna
- Ekologia
- Farmakologia
- Fizyka
- Inżynieria środowiskowa
- Medycyna
- Mikrobiologia
- Technologia chemiczna
- Zarządzanie projektami
- Badania kliniczne i przedkliniczne




Znaczenie inhibicji leucyloaminopeptydaz w opracowaniu nowoczesnych terapii
Wstęp
Leucyloaminopeptydazy to metalo-zależne proteazy, które hydrolizują N-końcową resztę neutralnego aminokwasu pochodzącego z białek. Enzymy te pełnią kluczową rolę w aktywacji, modyfikacji oraz degradacji protein. Nadczynność leucylo- aminopeptydaz jest odpowiedzialna za powstawanie stanów zapalnych, nowotworów oraz wzrost skłonności do infekcji wirusem HIV. Ze względu na funkcje, jakie leucylo- aminopeptydazy pełnią w organizmie, istotna jest regulacja pracy tych enzymów. Jedną z metod kontroli pracy leucyloaminopeptydaz jest ich inhibicja przez selektywnie działające cząsteczki inhibitorów. Zahamowanie aktywności tych enzymów może okazać się kluczem w opracowaniu nowej generacji leków [1].
Charakterystyka leucyloaminopeptydaz
Leucyloaminopeptydazy są to enzymy należące do grupy egzopeptydaz. Katalizują one hydrolizę hydrofobowego łańcucha na końcu N reszt aminokwasów pochodzących z peptydowych i białkowych substratów. Głównymi substratami leucyloaminopeptydaz są proteiny posiadające na końcu aminowym aminokwas z resztą boczną leucyny. Jednak specyficzność tych enzymów jest znacznie szersza. Wykazują one także wysoką aktywność w przypadku hydrolizy białek z N-końcową metioniną, fenyloalaniną lub izoleucyną. Natomiast słabymi substratami są białka posiadające na końcu N małe lub negatywnie naładowane reszty aminokwasowe jak: glicyna, alanina, seryna, walina oraz kwasy asparaginowy i glutaminowy. Leucyloaminopeptydazy wykazują enancjoselektywność i rozcinają wiązanie peptydowe jedynie L-aminokwasów. Ponadto są one metaloproteinazami, które do swej aktywności potrzebują jonów metali [2].
Struktura leucyloaminopeptydaz określona na podstawie badań krystalicznych, wykazała, że enzymy te występują w postaci homoheksameru składającego się z dwóch trimerów. Struktury peptydaz różnych przedstawicieli organizmów prokariotycznych i eukariotycznych są bardzo zbliżone. Leucyloaminopeptydaza wyizolowana z P. putida występuje w postaci heksameru składającego się z dwóch domen ze strukturami α i β. Domena końca C zawiera miejsce aktywne enzymu [3].
Leucyloaminopeptydazy są szeroko rozpowszechnione w przyrodzie. Aktywność tych enzymów została wykryta w organach i tkankach ssaczych, płynach ustrojowych oraz ścianach komórkowych, a także w organizmach niższych. Leucyloaminopeptydazy odgrywają istotną rolę w wielu procesach fizjologicznych, jak aktywacja i degradacja białek, dojrzewanie białek oraz metabolizm biologicznie aktywnych białek. Ponadto są one odpowiedzialne za prezentacje antygenów, regulacje poziomu hormonów, przemianę glutationu w wątrobie, angiogeneze, przekazywanie bólu, regulacje ciśnienia krwi czy kontrole proliferacji komórek [4]. Odpowiedniki tych enzymów ekspresjonowane przez Plasmodium falciparum biorą udział w ostatnich stadiach degradacji hemoglobiny, co jest odpowiedzialne za kliniczne objawy malarii [5].
- Cytozolowa leucyloaminopeptydaza
Cytozolowa leucyloaminopeptydaza (LAP) jest jedną z najlepiej poznanych aminopeptydaz, biorąc pod uwagę sekwencję, strukturę i mechanizm działania. Określono strukturę krystalograficzną cytozolowej leucyloaminopeptydazy wyizolowanej z soczewki wołowej. Enzym ten występuje w postaci heksameru zbudowanego z sześciu identycznych podjednostek, składających się z 487 aminokwasów. Cytozolowa leucyloaminopeptydaza posiada sześć miejsc aktywnych. W każdym z nich znajdują się dwa jony cynku (Zn488 i Zn489), które uczestniczą w procesie katalitycznym, bezpośrednio oddziałując z poszczególnymi elementami substratu. Oprócz tego poza miejscem aktywnym znajduje się jeden jon cynku (Zn490), który pełni rolę strukturalną. Jony cynku są koordynowane przez odpowiednie grupy karboksylowe reszty bocznej kwasów asparaginowego i glutaminowego. W natywnym enzymie jony cynku są połączone mostkiem, utworzonym przez cząsteczkę wody [6].
Cytozolowa leucyloaminopeptydaza jest odpowiedzialna za hydrolizę N-końcowego aminokwasu w substratach oligopeptydowych. Jej działanie jest ukierunkowane na hydrofobowe reszty alifatycznych i aromatycznych aminokwasów, takich jak leucyna czy fenyloalanina. Aktywność cytozolowej leucyloaminopeptydazy jest zależna od dwóch kationów metalu obecnych w miejscu aktywnym [2].
Aktywność cytozolowej leucyloaminopeptydazy została wykryta w wielu tkankach i organach ssaczych, w organizmach jednokomórkowych a także w roślinach. Podobnie jak wszystkie aminopeptydazy, cytozolowa leucyloaminopeptydaza pełni istotną rolę w fizjologicznych procesach aktywacji, modulacji i degradacji bioaktywnych peptydów. Ponadto uczestniczy ona w przemianach glutationu w wątrobie, regulacji poziomu hormonów oraz procesuje peptydy antygenowe, będąc mediatorem odpowiedzi immunologicznej organizmu. Zwiększony poziom cytozolowej leucyloaminopeptydazy obserwowany jest w różnych stanach patologicznych, jak: katarakta oka, stany zapalne, choroby nowotworowe, białaczka szpikowa czy wczesne stadia infekcji wirusem HIV. Potencjalne inhibitory cytozolowej leucyloaminopeptydazy budzą duże zainteresowanie, jako ewentualne środki przeciwnowotworowe [4].
Przeprowadzone badania wykazują, że cytozolowa leucyloaminopeptydza ekspresjonowana przez Plasmodium falciparum katalizuje uwolnienie neutralnych aminokwasów w ostatnim stadium degradacji hemoglobiny. Niszczenie erytrocytów gospodarza jest bezpośrednio odpowiedzialne za kliniczne objawy malarii. Zastosowanie odpowiednich inhibitorów, hamujących aktywność leucyloaminopeptydaz, może pełnić kluczową rolę w opracowaniu nowej generacji leków przeciwmalarycznych [5].
- Mikrosomalna leucyloaminopeptydaza
Mikrosomalna leucyloaminopeptydaza (APN) jest metalozależną egzopeptydazą o specyficzności podobnej do cytozolowej aminopeptydazy. Zazwyczaj występuje ona w postaci transmembranowej (CD13) oraz w formie rozpuszczalnej. Mikrosomalna leucyloaminopeptydaza jest niekowalencyjnym homodimerem, zbudowanym z identycznych podjednostek, składających się z 967 aminokwasów. Wewnątrzkomórkowy N-końcowy fragment tego enzymu jest zakotwiczony w błonie częścią heliakalną. Natomiast pozakomórkowa część C-terminalna zawiera miejsce aktywne enzymu i jeden jon cynku, który uczestniczy w procesie katalitycznym oraz inhibicji. Forma rozpuszczalna mikrosomalnej leucyloaminopeptydazy nie posiada części transmembranowej i została zidentyfikowana w plazmie, surowicy i moczu [7].
Mikrosomalna leucyloaminopeptydaza została wykryta u ssaków w komórkach nerwowych i nabłonkowych komórkach nerek. W układach biologicznych substratami APN są hormonalne peptydy oraz neuropeptydy. Mikrosomalna aminopeptydaza odgrywa istotną rolę w wielu procesach fizjologicznych związanych z: systemem odpornościowym, angiogenezą, tumorogenezą, przekazywaniem bólu, regulacją ciśnienia krwi czy końcowym procesowaniem peptydów generowanych przez proteazy trawienne. Biorąc pod uwagę udział APN w regulacji ciśnienia krwi, stanowi ona potencjalny cel w leczeniu nadciśnienia tętniczego [8].
Mikrosomalna leucyloaminopeptydaza występująca w postaci transmembranowej (APN/CD13) jest markerem ostrej białaczki szpikowej. Pełni ona istotną rolę w rozwoju nowotworów poprzez regulowanie takich procesów, jak kontakt komórek, metabolizm peptydów regulatorowych różnych typów komórek (proliferacja i inwazja komórek) oraz regulacja angiogenezy. APN/CD13 może pełnić ważną rolę w opracowaniu nowych leków przeciwnowotworowych [9].
Inhibitory leucyloaminopeptydaz
Podwyższony poziom leucyloaminopeptydaz obserwowany jest w różnych stanach patologicznych. Ze względu na bardzo ważną rolę, jaką te peptydazy pełnią w organizmie, niezwykle istotna jest regulacja pracy tych enzymów. Zahamowanie aktywności leucyloaminopeptydaz poprzez zastosowanie efektywnych i selektywnych inhibitorów jest kluczem w opracowaniu nowoczesnych terapii [1].
Poznano wiele naturalnych i syntetycznych inhibitorów leucyloaminopeptydaz. Do najbardziej efektywnych inhibitorów tych peptydaz należą produkty naturalne takie jak bestatyna i amastatyna, produkowane przez różne szczepy Streptomyces (Rys.1). Związki te należą do wolno wiążących peptydowych inhibitorów. Uważane są za analogi stanu przejściowego hydrolizy wiązania amidowego. Bestatyna i amastatyna koordynują obecny w centrum aktywnym Zn489 poprzez atom azotu N-końcowej grupy aminowej oraz atom tlenu grupy hydroksylowej. Mechanizm inhibicji jest kompetencyjny. Polega on na tym, że inhibitor i substrat współzawodniczą o miejsce aktywne cząsteczki enzymu. Przyłączenie cząsteczki inhibitora przez leucyloaminopeptydaze uniemożliwia związanie substratu i odwrotnie. Powstały kompleks enzym-inhibitor jest enzymatycznie nieaktywny. Możliwa jest kontrola szybkości reakcji poprzez regulację stężenia inhibitora. Bentatyna posiada działanie immunostymulujące oraz przeciwnowotworowe, co jest wykorzystywane w terapii niedrobnokomórkowego raka płuc [10].
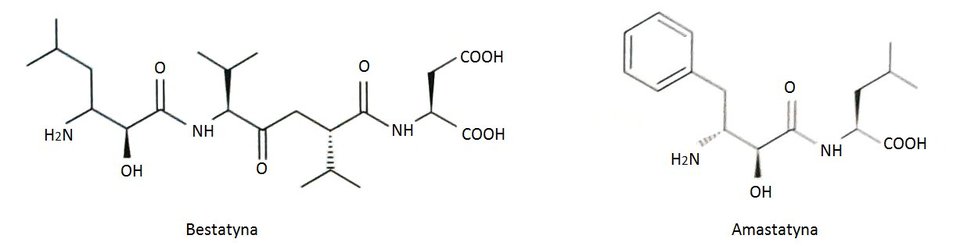
Rys.1. Wzory strukturalne naturalnych inhibitorów leucyloaminopeptydaz: bestatyny i amastatyny.
Otrzymano także wiele syntetycznych inhibitorów leucyloaminopeptydaz (Rys.2). Struktura większości z nich oparta jest na analogii do leucyny, w której grupę karboksylową zastąpiono inną, odpowiednio silnie oddziałującą z atomem cynku, np. aldehydową, tiolową, chlorometyloketonową, kwasu borowego czy hydroksamowego. Mechanizm inhibicji jest kompetycyjny. Polega on na tworzeniu oddziaływań podobnych do tych, które są obserwowane dla substratu, produktu lub wysokoenergetycznego stanu przejściowego reakcji katalizowanej przez enzym [10,11].
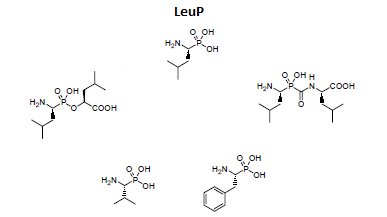
Rys.2. Przykłady syntetycznych fosforoorganicznych inhibitorów leucyloaminopeptydaz.
Do projektowania dalszych generacji inhibitorów wykorzystano fosfonowy analog L-leucyny (LeuP), jako strukturę wiodącą. Do jej zalet należy stabilność chemiczna, interesująca aktywność oraz selektywność wobec enzymu. Zastosowanie α-aminofosfonianów przyczyniło się także do zidentyfikowania zależności pomiędzy resztą P1 a specyficznością wobec LAP i APN (Rys.3). Cytozolowa leucyloaminopeptydaza (LAP) preferuje rozbudowany hydrofobowy podstawnik P1, natomiast mikrosomalna leucyloaminopeptydaza (APN), dodatkowy heteroatom. Znajomość struktury krystalicznej kompleksu LeuP z LAP pozwoliła na zaprojektowanie modyfikacji metodami modelowania molekularnego, a struktura LeuP umożliwia wydłużenie łańcucha poprzez dodanie nowej reszty P1' [10,11].
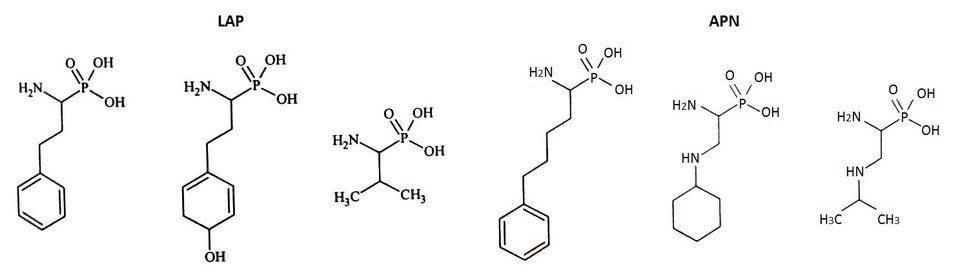
Rys.3. Wzory strukturalne fosforowych analogów aminokwasów z rozbudowaną resztą P1’.
Wprowadzenie tych zmian przyczyniło się do osiągnięcia jakościowego skoku efektywności inhibicji. Zaprojektowano nową klasę inhibitorów leucyloaminopeptydaz, analogów fosfinodipeptydów, zawierających grupę aminową w podstawniku P1′ (Rys.4). Związki te są odpornymi hydrolitycznie analogami stanu przejściowego reakcji katalizowanej przez enzym. Fosfinodipeptydy są najaktywniejszymi fosforoorganicznymi efektorami LAP ze stałymi inhibitorami rzędu nanomolarnego. Ponadto, fosfinowy analog z dodatkową grupą hydroksylową w pozycji P1' okazał się bardzo atrakcyjnym wobec APN [10,12].
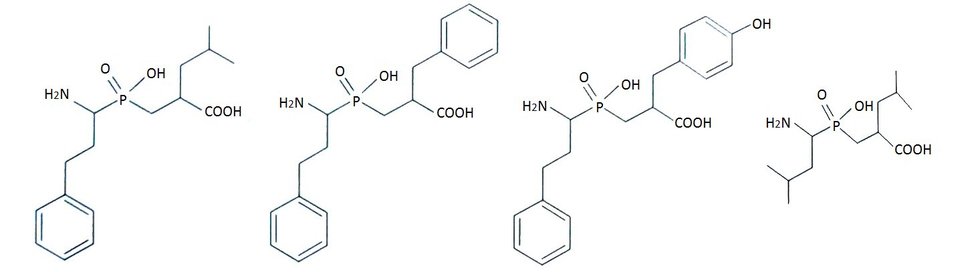
Rys.4. Wzory strukturalne fosfinowych pseudodipeptydów wobec LAP oraz APN.
Fosfinodipeptydy zostały zastosowane w badaniach nowych potencjalnych terapii przeciwmalarycznych. Wykazano, że są one skutecznymi inhibitorami rozwoju linii komórkowych in vitro, w tym odpornych na działanie standardowych leków, takich jak chlorochina. W badaniach in vivo fosfinodipeptydy hamowały rozwój nieletalnego modelu malarii u myszy. Hamowanie wybranych leucyloaminopeptydaz stanowi skuteczną strategię przeciwdziałania tej chorobie tropikalnej [5,10].
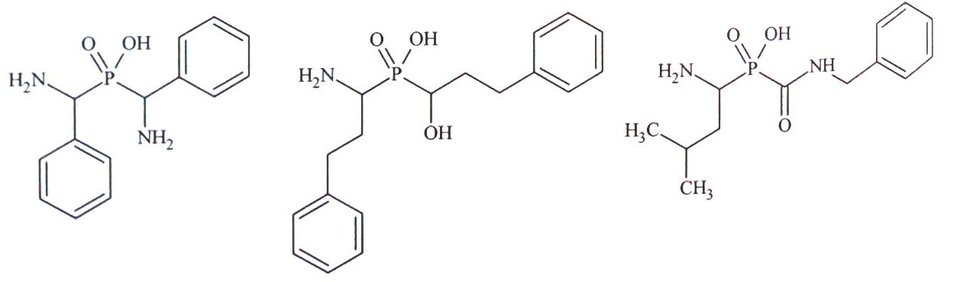
Rys.5. Przykłady fosfinowych analogów kwasu hydroksamowego.
Kolejną grupą inhibitorów leucyloaminopeptydaz są kwasy fosfonowe będące formalnymi analogami kwasu hydroksamowego. Związki te zawierają atom fosforu z niepeptydowym fragmentem P1’ oraz dwufunkcyjnym systemem chelatującym. Fosfinowe analogi kwasu hydroksamowego są bardzo dobrymi inhibitorami mikrosomalnej leucyloaminopeptydazy (APN) z IC50 w zakresie nanomolarnym (Rys. 5) [10,13].
Podsumowanie:
Poszukiwania nowych terapeutyków skupiają się na znalezieniu szlaków charakterystycznych dla procesów chorobotwórczych, a także enzymów odpowiedzialnych za rozwój danej choroby, a następnie obniżeniu ich aktywności poprzez zastosowanie odpowiednich inhibitorów. Wyzwaniem dla naukowców jest opracowanie selektywnych inhibitorów leucyloaminopeptydaz, które mogą pełnić ważną rolę w opracowaniu nowej generacji leków.
W cząsteczkach potencjalnych inhibitorów leucyloaminopeptydaz wymagana jest obecność grupy aminowej, pierścienia fenylowego oraz grupy fosfonianowej. Istotne znaczenie dla aktywności tych inhibitorów odgrywa grupa fosfonianowa, która kompleksuje jony cynku obecne w miejscu aktywnym enzymów. Analogi aminokwasów i peptydów zawierające atom fosforu są obecnie intensywnie badanymi syntetycznymi inhibitorami leucyloaminopeptydaz. Optymalizacja struktury P1, modyfikacja cynkowego systemu chelatującego oraz regionu Pn' pozwoliła na znalezienie związków efektywnie hamujących aktywność aminopeptydaz. Prekursorami tych związków mogą być pochodne azyrydynowe, które odgrywają istotną rolę w syntezie organicznej, ze względu na wysoką regio- i stereoselektywność reakcji z ich udziałem. Związki zawierające w swej strukturze pierścień azyrydynowy wykazują także aktywność biologiczną i stosowane są jako antybiotyki czy leki przeciwnowotworowe.
Autor: Katarzyna Czuba
Literatura:
- Taylor A. Aminopeptidases: structure and function. FASEB J. 1993. 7, 290-298.
- Matsui M., Fowler J. H., Walling L. L. Leucine aminopeptidases: diversity in structure and function. Biological Chemistry Hoppe-Seyler. 2006. 387. 12, 1535-44.
- Pícha J., Liboska R., Buděšínský M., Jiráček J., Pawełczak M., Mucha A. Unusual activity pattern of leucine aminopeptidase inhibitors based on phosphorus containing derivatives of methionine and norleucine. Journal of Enzyme Inhibition and Medicinal Chemistry. 2011. 26. 2, 155-61.
- Taylor A. Aminopeptidase: towards a mechanism of action, Trends Biochem. Sci. 1993. 18, 167-171.
- Skinner-Adams T.S., Stack C.M., Trenholme K.R., Brown C.L., Grembecka J., Lowther J., Mucha A., Drag M., Kafarski P., McGowan S., Whisstock J.C., Gardiner D.L., Dalton J.P. Plasmodium falciparum neutral aminopeptidases: new target for anti-malarials. Trends Biochem. Sci. 2010. 35, 53-61.
- Sträter N., Lipscomb W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: active site solvent structure and binding mode of L-leucinal, a gemdiolate transition state analog, by X-ray crystallography. Biochem. 1995. 34, 14792-14800.
- Luan Y., Xu W. The structure and main functions of aminopeptidase N. Curr. Med. Chem. 2007. 14, 639-647.
- Bank U., Bohr U.R., Reinhold D., Lendeckel U., Ansorge S.. Malfertheiner P., Tager M. Inflammatory bowel diseases: multiple benefits from therapy with dipeptidyl and alanylaminopeptidase inhibitors. Front Biosci. 2008.13, 3699-3713.
- Bhagwat S.V., Lahdenranta J., Giordano R., Arap W., Pasqualini R., Shapiro L.H. CD13/APN is activated by angiogenic signals and is essential for capillary tube formation. Blood. 2001. 97, 652-659.
- Węglarz E. Leucyloaminopeptydazy – znaczenie i strategie inhibicji. Na pograniczu chemii i biologii. 2010. Tom XXV. 81-89.
- Drąg M., Grembecka J., Pawełczak M., Kafarski P. α-Aminoalkylphosphonates as a tool in experimental optimisation of P1 side chain shape of potential inhibitors in S1 pocket of leucine- and neutral aminopeptidases. Eur. J. Med. Chem. 2005. 40, 764-771.
- Grembecka J., Mucha A., Cierpicki T., Kafarski P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003. 46, 2641-2655.
- Drag M., Grzywa R., Oleksyszyn J. Novel hydroxamic acid-related phosphinates: inhibition of neutral aminopeptidase N (APN). Bioorg. Med. Chem. Lett. 2007. 17, 1516-1519.
- http://pubs.acs.org/doi/abs/10.1021/jm3002058
Recenzje