Streszczenie
Tradycyjne badania, szczególnie badania białek przeprowadzano in vivo (wenątrz żywego organizmu) bądź in vitro, czyli w probówce. Obecnie rozwija się nowy typ badań in silico. Pozwala on badać białka (strukturę, właściwości i występowanie) bez użycia metod doświadczalnych.
Wprowadzenie
In silico jest wykorzystywane w znaczeniu "wykonywane na komputerze lub za pomocą symulacji komputerowej”. Zwrot ten został wprowadzony w 1989 roku poprzez analogię do wyrażeń in vivo i in vitro oraz in situ. Zwroty te są powszechnie stosowane w biologii jednak nowy termin jest przypisany chemii, mimo, że poraz pierwszy zwrot ten został użyty przez biologów [1]. Pierwsze zastosowania także dotyczyły ściśle biologii, szczególnie biologii komórki i biologii molekularnej. Jednak obecnie zwrot "in silico" używany jest do określenia symulacji komputerowych i modelowania procesów fizycznych i chemicznych, w szczególności do badań nad cząsteczkami o dużej masie cząsteczkowej, w tym białek. Obecnie dostępne są dane o sekwencji białek i ich struktur. Dlatego też biochemicy, zajmujący się zagadnieniami związanymi z białkami co raz częściej korzystają z komputera a niżeli z doświadczeń empirycznych. Ogromny zakres danych przechowywany jest w bazach danych a praca z nimi zwana jest bioinformatyką.
Historia bioinformatyki białek
Pierwszą bazą danych dotyczących biochemicznych związków o dużej masie cząsteczkowej był Zbiór Informacji o Białkach (PIR). Nazwa wskazywałaby na to, że dane tam zebrane dotyczyły przede wszystkim sekwencji aminokwasów. PIR został założony przez Margaret Dayhoff.
Podstawowe zagadnienia bioinformatyki [2]
1. Katalogowanie informacji biologicznych
- b. wyszukiwanie sekwencji,
2. Analiza sekwencji DNA
- c. wyszukiwanie sekwencji kodujących,
- d. wyszukiwanie sekwencji regulatorowych
- e. wyszukiwanie sekwencji repetytywnych,
3. Analiza sekwencji
4. Ustalanie ewolucyjnych relacji:
- a. pomiędzy zbiorami sekwencji
- c. tworzenie drzew filogenetycznych
5. Genotypowane
- a. wyszukiwania genów odpowiedzialnych za choroby genetyczne,
6. Analiza ekspresji genów (głównie analiza danych z mikromacierzy)
7. Analiza sekwencji białek, nazywana też proteomiką
- a. porównywanie sekwencji,
- b. wyszukiwanie domen i motywów,
- c. przewidywanie własności fizyko-chemicznych,
- d. drugo- i trzecio-rzędowej struktury białka,
- e. lokalizacja w obrębie komórki,
- f. analiza danych z eksperymentow spektroskopwych)
8. Katagolowanie
- b. analiza dróg metabolicznych (np metabolizm lipidów)
- c. analiza dróg sygnałowych (np od receptora na powierzchni komórki poprzez kaskadę kinaz do czynników transkrypcyjnych)
9. Modelowanie układów biologicznych
10. Wirtualne dokowanie
11. Komputery DNA
12. Morfometria / analiza obrazu
Poszukiwanie struktury białkaGłówną metodą poszukiwania strukturu białka jest ab initio. Ab initio opiera się na hipotezie „termodynamicznego” zwijania białek (natywna struktura sekwencji białkowej odpowiada stanowi globalnego minimum energetycznego). Metoda ta ma wiele utrudniem jednym z nich jest paradoks Liventhala, czyli liczba konformacji swobodnego łańcucha dąży do nieskończoności. A zatem jeśli liczba możliwych stopni swobody jednego aminokwasu wynosiłaby 2, to dla łańcucha N=150 aminokwasów liczba możliwych konformacji wynosi 2150, czyli, wiedząc, że czas sprawdzania jednej konformacji wynosi 10-13 s, to dla takiego białka sprawdzenie wszystkich stanów wynosi:2150×10-13 s = 4.6×1024roku (czas „życia‟ Ziemi = 4.5×109lat)

Tab.1 Rodzaje badań struktury białek
Aby móc stworzyć obraz przestrzenny białka należy stworzyć: model molekuły oraz model oddziałowywań międzyatomowych (oddziaływania przez wiązania kowalencyjne (Bonded Interactions ); Oddziaływania bez wiązań (Non-bonded Interactions): hydrofobowe, Van der Waalsa, elektrostatyczne, wiązania wodorowe). Przy projektowaniu struktury białka stosuje zasady mechaniki Newtona (uproszczenie mechaniki kwantowej). Po stworzeniu molekuły należy zastosować tzw. regułę minimum: zmiana konformacji molekuły ma doprowadzić do zmniejszenia jej energii wewnętrznej. Celem jest absolutne zero (energia kinetyczna = 0). Otrzymuje się przy zastosowaniu dynamiki molekularnej. Metody optymalizujące:
– rozwiązanie równań Newtona
– próbkowanie przestrzenie stanów (MC)
– algorytm genetyczny
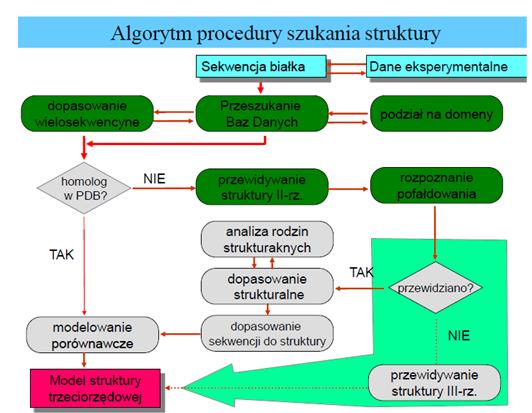
Rys. 1 Algorytm procedury szukania struktury białka [2]
Szukanie podobieństw pomiędzy strukturami białek [2]W celu znalezienia bądź porównania struktury białek stosuje się metodę LGA (Local-Global Alignment) metoda szukania podobieństwa 3D między strukturami białek. Metroda ta zawiera dwie procedury:
– LCS (Longest Continuous Segments) loaklizuje najdłuższe fragmenty struktury, które pasują do siebie z RMSD poniżej zadanej wartości
– GDT (Global Distance Test) szuka najdłuższych, niekoniecznie ciągłych fragmentów „równoważnych” aminokwasów, które są w odległości niewiększej niż zadana.
Bazy danych – białka [3]Bazy danych oprócz danych strukturalnych i tworzenia z tych danych magazynów, również weryfikują wprowadzane dane (o ile to możliwe). Dodatkowo wprowadzone posiadaja komentarz o danym białku oraz najważniejsze hiperłącza do innych ważnych źródeł dotyczących danego białka. Wiekszość baz danych posiada wolne oprogramowanie (freeware) umożliwiając lub tylko ułatwiające przeglądani informacji. Większość analiz biochemicznych i chemicznych dotyczących białek można wykonać on-line. Zestawienie użytecznych programów przedstawione zostało w tabeli (Tab.1). Każdy z programów specjalizuje się w innej dziedzinie. Przykładem są:
a. Analiza białek – szwajcarski Instytut Bioinformatyki ExPASy
b. Struktura trójwymiarowa białek – Wirtualne Laboratorium Badawcze Bioinformatyki Strukturalnej na uniwersytecie Rutgers (potocznie zwane bazą danych z Brookhaven)
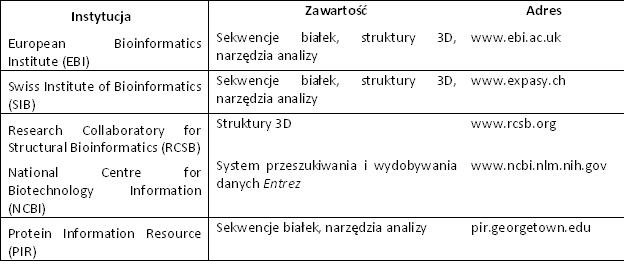
Tab. 2. Wybrane bazy danych o dużym znaczeniu w chemii białek [3]
Inne zastosowania badań in silicoBadania in silico we wszystkich naukach przyrodniczych pozwalają projektować procesy i cząsteczki przyspieszyć tempo odkryć, a jednocześnie zmniejszając koszty prowadzonych prac laboratoryjnych, jak i klinicznych. Dzięki badaniom in silico w 2010 roku, naukowcy odkryli potencjalny inhibitor karcenogenezy [4]. Badnia in silico pozwoliły na szybszy rozwój farmacji [5]. Nastąpił także rozwój mikrobiologii, dzięki możliwościom projektowania cyklów komórkowych [6]. Tworzenie baz danych genomu, sekwencji DNA odbywa się także za pomocą tej metody. Najważniejszymi osiągnięciami tej metody są:
- Analiza komórek prokariotycznych i eukariotycznych gospodarzy
- Optymalizacja bioprocesów
- Analiza, interpretacja i wizualizacja zbiorów danych z różnych źródeł np. genomu, transkryptomu, proteomu
Podsumowanie
Metoda in silico jest coraz częściej stosowana w badaniach naukowych. Metoda ta szybko się rozwija poszerzając znajomość struktur, właściwości i występowania białek. Należy pamiętać, że aby móc w pełni z niej korzystać należy posiadać komputer o odpowiedniej mocy obliczeniowej.
Literatura:
[1] Miramontes P. Un modelo de automaty Celular para la evolución de los ácidos nucleicos. 1992. UNAM
[2] http://www.staff.amu.edu.pl
[3] Doonan Sh. 2008. Białka i peptydy. Wydawnictwo Naukowe PWN
[4] Röhrig, Ute F.; Awad, Loay; Grosdidier, AuréLien; Larrieu, Pierre; Stroobant, Vincent; Colau, Didier; Cerundolo, Vincenzo; Simpson, Andrew JG et al. 2010. Journal of Medicinal Chemistry 53(3): 1172-1189
[5] www.sciencedaily.com
[6] Li S, Brazhnik P, Sobral B, Tyson JJ, 2009 Temporal Controls of the Asymmetric Cell Division Cycle in Caulobacter crescentus.Comput. Biol 5(8)
opracowała: Katarzyna Wójciuk
Tagi:
in silico,
bioinformatyka,
struktura białek,
De Novo Ab initio,
algorytm procedury szukania struktury białka,
lab,
laboratorium,
laboratoria
wstecz
Podziel się ze znajomymi









