
|

Zamknij X
|





Osocze krwi jest ważnym środkiem transportującym metabolity w organizmie ssaków, a analiza chemiczna surowicy może dostarczyć wielu informacji odnoszących się do stanu biochemicznego osobnika i jest ona istotna dla celów diagnostycznych. Osocze jest bardzo złożone pod względem fizykochemicznym, ponieważ składa się z szeregu organicznych i nieorganicznych składników o szerokim zakresie mas cząsteczkowych i klas chemicznych, co sprawia, że analiza osocza nie jest łatwa. Coraz częściej do analizy osocza krwi poza tradycyjnymi metodami wykorzystuje się spektroskopię NMR (spektroskopia magnetycznego rezonansu jądrowego), która dostarcza bardzo użytecznych w diagnostyce informacji jakościowych i ilościowych związanych z zaburzeniami metabolicznymi [16].
Wykrywanie kreatyniny
W środowisku zasadowym z kwasem pikrynowym kreatynina tworzy czerwono zabarwiony kompleks.
Wykonanie:
Do 2 ml przesączu po kwasie trichlorooctowym należy dodać po kilka kropli nasyconego roztworu kwasu pikrynowego oraz 10% roztwór wodorotlenku sodu (NaOH). W wyniku reakcji dochodzi do wytworzenia pomarańczowego zabarwienia roztworu [1].
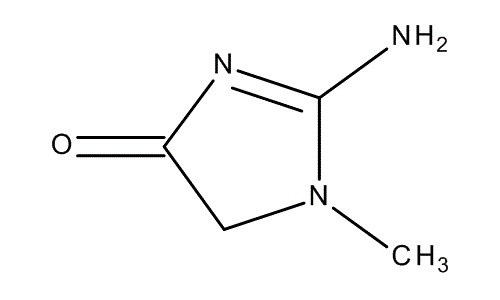
Zdjęcie: wzór kreatyniny, http://www.merckmillipore.com/PL/en/product/Creatinine,MDA_CHEM-105206, [7].
Odczyn Jaffe’go
1 ml roztworu kreatyniny zmieszać z 2 ml nasyconego roztworu kwasu pikrynowego i 0,5 ml 10% roztworu wodorotlenku sodu (NaOH). W wyniku reakcji powstaje barwny pomarańczowo-czerwony kompleks [18].
Oznaczanie kreatyniny metodą kolorymetryczną z wykorzystaniem kitu Cayman Chemical (Creatinine(serum) Colorimetric Assay Kit, wg protokołu ze strony: https://www.caymanchem.com/pdfs/700460.pdf)
Zestaw może być stosowany do oznaczania pomiaru stężenia kreatyniny w osoczu i surowicy krwi. Test opiera się na reakcji Jaffe’go, w której powstają barwne kompleksy (żółte/pomarańczowe), kiedy metabolit traktowany jest alkalicznym pikrynianem (sól kwasu pikrynowego). Wskaźnik wywołania zabarwienia jest wprost proporcjonalny do stężenia kreatyniny w próbce i mierzony na podstawie absorbancji między 490 – 500 nm. Kinetyczny charakter testu eliminuje zakłócenia pochodzące od zanieczyszczeń surowicy, takich jak lipidy czy bilirubina [17].
Przygotowanie próbek
Osocze: Pobrać krew z zastosowaniem antykoagulanta: heparyny lub cytrynianu. Następnie, odwirować krew przy 700 – 1000 x g przez 10 minut w 4°C. Pipetą zebrać żółtą warstwę osocza- uważając by nie naruszyć białej warstwy kożucha, który powstał po wirowaniu. Tak przygotowaną próbkę należy przechowywać na lodzie lub w temp. -80°C do momentu oznaczenia. Próbki osocza są stabilne przez okres 1 miesiąca. Osocze nie musi być rozcieńczane przed oznaczeniem.
Surowica: Pobrać krew bez użycia natykoagulanta, następnie zostawić krew do zakrzepnięcia na 30 minut w temperaturze 25°C. Próbkę krwi odwirować przy 2000 xg przez 15 minut w 4°C. Zebrać pipetą górną warstwę próbki (żółta warstwa) uważając, by nie naruszyć białej warstwy kożucha. Surowicę przetrzymywać na lodzie. Jeśli oznaczenie nie będzie wykonywane tego samego dnia próbke należy zamrozić w temperaturze -80°C- próbka będzie stabilna przez 1 miesiąc (należy unikać rozmrażania i ponownego jej zamrażania). Surowica nie musi być rozcieńczana przed oznaczeniem [17].
Procedura oznaczenia kreatyniny z uzyciem kitu
Oznaczenie wykonuje się na 96-dołkowej płytce, przy czym nie istnieje określony wzór nakładania na płytkę surowicy, standardu i próbki badanej. Ważne by każda próbk amierzona była w dwóch egzemplarzach (Creatinine(serum) Colorimetric Assay Kit, wg protokołu ze strony: https://www.caymanchem.com/pdfs/700460.pdf))
A-H = Standard
S1 – S40= przykładowe studzienki
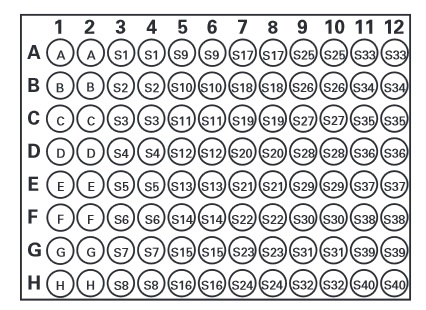
Zdjęcie: Rozmieszczenie prób badanych na płytce, zdjęcie ze strony producenta kitu (https://www.caymanchem.com/pdfs/700460.pdf)
Informacje ogólne dotyczące kitu
- całkowita objętość we wszystkich studzienkach wynosi 215 µl
- wszystkie odczynniki należy doprowadzić do temperatury pokojowej przed rozpoczęciem testu
- nie jest konieczne wykorzystanie wszystkich dołków na płytce w trakcie oznaczenia
- zalecane jest aby wzorce i próbki mierzone były w potórzeniach (zalecane 3 powtórzenia)
- test należy przeprowadzać w temperaturze pokojowej
- pomiar absorbancji w zakresie 490 – 500 nm [17].
Przygotowanie standardu:
Do oznaczenia kreatyniny w osoczu lub surowicy należy przygotować zgodnie z tabelą podaną w instrukcji producenta . Należy przygotowac 8 czystych, szklanych probówek opisanych kolejno od A-H, po czym odmierzyć do nich (zgodnie z instrukcją) standard kreatyniny (20 mg/dl) oraz wodę HPLC-grade (zdjęcie poniżej):
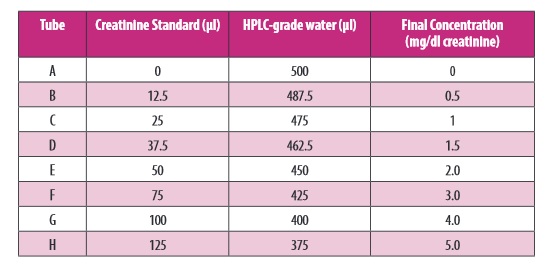
Zdjęcie: przygotowanie standardu kreatyniny, https://www.caymanchem.com/pdfs/700460.pdf [17].
Wykonanie oznaczenia:
Pomiar stężenia kreatyniny w reakcji z pikrynianem alkalicznym z wykorzystaniem gotowego zestawu odczynników [procedura z zestawu firmy BioSystems ze strony: http://chemklin.sum.edu.pl/uploaded/kreatynina.pdf
Kretynina w próbie badanej, w środowisku zasadowym ulega reakcji z pikrynianem, czego wynikiem jest powstanie barwnego kompleksu w próbce. W celu uniknięcia interferencji szybkośc tworzenia się kompleksu mierzona jest w krótkim czasie. Jako materiał do badań wykorzystuje się surowicę, osocze lub mocz, które pobierane są wg standardowych procedur. Świeży mocz należy rozcieńczyć pzred oznaczeniem wodą destylowaną (w stosunku 1:50). Kreatynina w próbie badanej jest stabilna przez 24 godziny ( w temperaturze 2-8°C) [22].
Odczynniki dołączone do zestawu:
Odczynnik A: wodorotlenek sodu (0,4 mol/l)
Odczynnik B: kwas pikrynowy (25 mmol/l)
Standard S: standard glukozy/mocznika lub kreatyniny: glukoza 100mg/dl, mocznik 50mg/dl, kreatynina 2mg/dl (177µmol/l) [22].
Wykonanie oznaczenia:
Na podstawie otrzymanych wyników obliczyć stężenie kreatyniny wg wzoru:
(A2 – A1) próby/ (A2 – A1) standardu x C standardu x współczynnik rozcieńczenia próby = C próby
Jeżeli do kalibracji używa się standardu kreatyniny dołączonego do zestawu, w obliczeniach stężenia należy wprowadzić inne wartości wspólczynnika rozcieńczenia próby, tj.:
Dla próbki surowicy i osocza: x 2 = mg/dl kreatyniny
x 177 =µmol/l kreatyniny
Dla próbki moczu: x 100 = mg/dl kreatyniny
x 8840 = µmol/l kreatyniny [22].
Metoda enzymatyczna do oznaczania poziomu kreatyniny w surowicy krwi, osoczu lub moczu (http://www.wiener-lab.com/wiener/catalogo/archivos/6322_creatinina_enzimatica_aa_liquida_pl.pdf)
Kreatynina zaliczana jest do substancji wysoce rozpuszczalych. W większości związek ten eliminowany jest z organizmu przez filtrację nerkową. Wykrywanie kreatyniny w surowicy krwi jak również oznaczanie endogennego klirensu kreatyniny są ważnymi badaniami w diagnostyce wielu chorób nerek. Klirens kreatyniny jest parametrem odzwierciedlającym wielkość przesączania kłębuszkowego (tzw. GFR). Jego oznaczanie jest przydatne w ocenie funkcji nerek [19],[20].
Testu oceny wielkości przesączania kłębuszkowego (GFR) wykonywany jest w trakcie stosowania leków nefrotoksycznych. Oznaczanie klirensu kreatyniny jest podstawowym badaniem czynności nerek. Wskaźnik ten określa zdolność nerek do oczyszczania krwi (osocza) z kreatyniny. Aby wykonać to badanie konieczne jest oznaczenie stężenia kreatyniny w dobowej zbiórce moczu (określanej skrótem: DZM) oraz w surowicy krwi.
Stężenie kreatyniny w osoczu zależy o kilku czynników, w tym od masy mięśniowej, wieku, ilości wody w przestrzeni pozakomórkowej oraz od przesączania kłębuszkowego. Jak już wspomniano kreatynina jest prawie wyłącznie wydalana przez nerki. Jednakże, w przypadku znacznego ograniczenia filtracji nerkowej jest ona także wydzielana przez cewki nerkowe.
W wyniku różnych chorób nerek, wraz z utratą czynnych nefronów dochodzi do obniżenia filtracji, a tym samym klirensu. W zespołach nerczycowych i ciężkich niewydolnościach nerek wartość klirensu kreatyniny jest zawyżona, co ma związek z wydzielaniem cewkowym. Wtedy też utrudnione jest prawidłowe określenie wartości GFR. Należy mieć też na uwadze, że zwiększony klirens występuje fizjologicznie u kobiet w ciąży [20].
Zasada działania testu WienerLab oparta jest na kilku reakcjach, w wyniku których powstaje barwnik cholinoiminowy. Intensywnośc koloru tego barwnika jest wprost proporcjonalna do stężenia kreatyniny w badanej próbce [19]. W trakcie oznaczenia zachodzą poniższe reakcje:
Kratyninaza
Kreatynina + woda → kreatyna
Kreatynaza
Kreatyna + woda → sarkozyna + mocznik
Oksydaza sarkozyny
Sarkozyna + woda + tlen → glicyna + aldehyd mrówkowy + nadtlenek wodoru
Peroksydaza
Nadtlenek wodoru + 4-aminopirydyna +TOOS (N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-metyloanilina) → barwnik cholinoiminowy [19].
Odczynniki zestawu:
Odczynnik A: roztwór zawierający 36 kU/l kreatynazy,11 kU/l oksydazy sarkozyny, 300 kU/l katalazy, 3kU/l oksydazyaskorbinowej i 20 mmol/l buforu Good‘a pH 8,2 z 1 mmol/l N-ethyl-N-(2-hydroxy-3 sulfopropyl)-3-metyloaniliny (TOOS)
Odczynnik B: oztwór zawierający 4 mmol/l 4-aminofe-nazonu (4-AP), 370 kU/l kreatyninazy, 15 kU/l peroksydazy,0,8 g/l azydku sodu oraz 20 mmol/l buforu Good‘a pH 8,0
Standard (próba wzorcowa S): 20 mg/l roztworu kreatyniny
Materiał do badań może stanowić surowica, osocze lub mocz. W przypadku przeprowadzania oznaczenia na próbce surowicy lub osocza należy je pobrać w klasyczny sposób, z tym, że w przypadku osocza wskazane jest użycie heparyny jako antykoagulantu. Do oznaczenia używa się zbiórkę moczu 2 godzinną lub 24 godzinną, mocz przechowywać w temperaturze 2-10°C podczas zbiórki. Obliczyć diurezę (oceniana na podstawie pomiarów objętości moczu, który został wydalony w określonym czasie, np. w ciągu doby lub godziny tj. diureza dobowa, diureza godzinowa, przy równoczesnym uwzględnieniu ilości podanych płynówi ), do badania wziąć podwielokrotność i wykonać rozcieńczenie 1:50 z użyciem wody destylowanej. W przypadku 2-godzinnej zbiórki moczu należy pomnożyć objętość przez 12 w celu obliczenia wydalania kreatyniny w ciągu 24 godzin [19].
Osocze lub surowica powinna zostać oddzielona od komórek w ciągu 2 godzin od pobrania. Próbki te mogą być przechowywane w temp. 2-10°C do trzech dni (należy je zabezpieczy przed światłem). Próbkę moczu można przechowywać w temperaturze 2-10°C do 4 dni bez dodatkowego konserwowania [19].
Wykonanie: oznaczenie należy wykonywać w temperaturze 37°C przy długości fali równej λ=546 nm.
Wykonanie obliczeń na podstawie uzyskanych pomiarów:
Kreatynina w surowicy (mg/l)= [(U2 – B2) – (U1 – B1) x k) x f
f= 20 mg/l / (S2 – B2) - (S1 – B1) x k, gdzie:
k= obj. próby ślepej (ml) / końcowa objętość (ml) = 2,57 ml / 3,82 ml = 0,673
Kreatynina w moczu (g/24h)= Kreatynina (mg/l) x 50 x U/ 1000 = kreatynina (mg/l) x U /20
gdzie:
U - objętość diurezy wyrażona w litrach/ 24 godziny
50 - współczynnik rozcieńczenia
1000 - konwersja mg na gramy [19].
Endogenny klirens kreatyniny (E.C.C)
E.C.C (ml/min)= kreatynina w moczu (g/24 godz)/ kreatynina w surowicy (mg/l) x 694 ml/min
gdzie:
694ml/min= (g/24 godz) / (mg/l)= 1000 mg x 1000 ml/ 1 mg x 1440 min= 1 000 000 ml/ 1440 min [19].
W 1980 roku Fossati P. i Prencipe L. przedstawili pracę, w której opisali ulepszony system wykrywania chromogenu, który w połączeniu z enzymatycznym utlenianiem kwasu moczowego prowadził do bezpośredniego oznaczania stężenia kwasu moczowego w płynach biologicznych. Opracowana metoda była niezawodna, prosta i szybka, a ponadto nadawała się do ręcznych i automatycznych procedur oznaczania. Wprowadzona metoda nie była pierwszą na rynku, już wcześniej istniały podobne metody, jednakże posiadały one sporo wad m.in. wymagały długich czasów inkubacji , przez co wydłużał się całkowity czas wykonania oznaczenia, a także pojawiały się fałszywie ujemne lub dodatnie wyniki, które były skutkiem obecności w próbce substancji zakłócających [21]. Reakcja sprzęgania pomiędzy fenolem i 4-aminofenazonem dająca czerwony chromogen chinonoiminy była znana od dawna i powszechnie stosowana w chemii klinicznej od czasu kiedy Trinder P. (1969) zaczął ją stosować do enzymatycznego oznaczania glukozy [21]. Na podstawie takiego zastosowania badacze przypuszczali, że podobną reakcję można zastosować do pomiaru stężenia kwasu moczowego. Jednakże, wraz z rozpoczęciem prac pojawiły się trudności wynikające z niskiego stężenia kwasu moczowego w surowicy i niezgodnościami pomiędzy pH roboczej peroksydazy chrzanowej i pochodzącej od zwierząt urykazy. Ponadto, układ chromogenowy Emerson-Trinder miał znaczną wadę, a mianowicie reakcja oksydacyjnego sprzęgania przebiegała nieprawidłowo w obecności bilirubiny i innych związków redukujących. Wada ta powoodwała zawyżenie koloru reakcji, a tym samym fałszywy wzrost stężenia substancji mierzonej [21]. Interferencja wywołana przez związki redukujące (takie jak kwas askorbinowy) wynika głównie ze współzawodnictwa z chromogenem nadtlenku wodoru (w reakcji katalizowanej przez peroksydazę) w momencie powstawania koloru w reakcji [21].
Interferencja wywołana przez bilirubinę jest przeszkodą w ustaleniu metabolitów w surowicy krwi w metodzie Trindera (1969) z chromogenem, a zatem jest to główna wada w momencie, gdy analizowane są próbki zawierające wysokie stężenia bilirubiny [21]. Ciągłe badania nad ulepszeniem metody doprowadziły do wprowadzenia kilku jej ulepszeń, m.in. zastosowano bakteryjną urykazę produkowaną przez bakterie Aspergillus flavus, która nie traci aktywności w pH maksymalnej aktywności peroksydazy chrzanowej. Dodatkowo do metody wprowadzono żelazocyjanki, dzięki którym uniknięto ewentualnych zakłóceń pochodzących od wielu substancji takich jak bilirubina. Widmo barwnika otrzymanego w reakcji mierzono przy 520 nm, chemiczne zakłócenia eliminowano przez wprowadzenie żelazocyjanków do stosowanych wóczas odczynników [21].
W 1980 roku zautomatyzowaną metodę wprowadzono do powszechnego użytku. Wprowadzona powyższa jednoetapowa metoda została tak zaprojektowana, by można było wykonywać ją na wielu różnych automatycznych urządzeniach. W ciągu roku od wprowadzenia metoda została zastosowana na ponad 50 różnych przyrządach laboratoryjnych. Z powodu wielu zalet, chromogenna metoda Fossat P. i wsp. została przyjeta do pomiaru innych ważnych analitów, takich jak triglicerydy czy kreatynina, które to w reakcji z enzymami generują nadtlenek wodoru. Zasada oznaczania (z niewielkimi zmianami w skałdzie odczynników) jest nadal powszechnie stosowana w badaniach [21].
Autor: Lidia Koperwas
Literatura:
[1]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s.580-582, 592-595.
[2]. http://www.ibmb.uni.wroc.pl/studia/2.pdf
[3]. Rodriguez-Vico F., Martinez-Cayuela M., Zafra M.F., Garcia-Peregrin E., Ramirez H., 1991. Lipids 26 (77-80).
[4]. Niebiałkowe związki azotowe -mocznik, kwas moczowy, kreatynina; ilościowe oznaczanie kreatyniny metodą Folina-Wu. Ćwiczenie nr 13. http://farmacja.cm-uj.krakow.pl/public_includes/upload/bioch_farm_cwiczenie_nr_13.pdf
[5]. Posadzy-Małaczyńska A., Tykarski A., 2011. Kwas moczowy w chorobach sercowo-naczyniowych – co nowego?. Borgis - Postępy Nauk Medycznych s3/2011, s. 46-50. http://www.pnmedycznych.pl/shown.php?ktory=4061
[6]. Drabczyk R., 2014. Kreatynina. Medycyna praktyczna dla pacjentów. http://nefrologia.mp.pl/diagnostyka/show.html?id=51973
[7]. http://www.merckmillipore.com/PL/en/product/Creatinine,MDA_CHEM-105206
[8]. http://www.merckmillipore.com/INTL/en/product/poland/chemicals/mocznik,MDA_CHEM-108486
[9]. Majdan M., Borys O., 2010. DNA i schorzenia towarzyszące podwyższonemu stężeniu kwasu moczowego. ANNALES ACADEMIAE MEDICAE STETINENSIS ROCZNIKI POMORSKIEJ AKADEMII MEDYCZNEJ W SZCZECINIE 2010, 56, SUPPL. 1, 34–39. http://www.pum.edu.pl/__data/assets/file/0009/29817/SUPLEMENT_56-01_05.pdf
[10]. Januszkiewicz-Caulier J., Franek E., 2011. Kwas moczowy w chorobach nerek, serca i naczyń. 31
K L I N I C Z N A I N T E R P R E T A C J A W Y N I K Ó W B A D A Ń. Choroby Serca i Naczyń 2011, tom 8, nr 1, 31–37. http://czasopisma.viamedica.pl/chsin/article/viewFile/18633/14657
[11]. Oznaczanie mocznika w płynach ustrojowych metodą hydrolizy enzymatycznej, http://pchba.amu.edu.pl/cw%20CBA/cw3.pdf
[12]. Ayelet Erez MD, PhD, 2013. Argininosuccinic aciduria: from a monogenic to a complex disorder. Genetics in Medicine (2013) 15, 251–257, http://www.nature.com/gim/journal/v15/n4/fig_tab/gim2012166f3.html.
[13]. http://chemklin.sum.edu.pl/uploaded/Chemia%20kliniczna/Kwas%20moczowy.pdf
[14]. Hunter G., 1957. A method for deproteinization of blood and other body fluids. J.clin. Path. (1957), 10, 161.
[15]. Sakuma R., Nishina T., Kitamura M., 1987. Deproteinizing Methods Evaluated for Determination of Uric Acid in Serum by Reversed –Phase Liquid Chromatography with Ultraviolet Detection. Clin. Chem. 33 No 8, 1427-1430 (1987). [16]. Daykin CA, Foxall PJ, Connor SC, Lindon JC, Nicholson JK. The comparison of plasma deproteinization methods for the detection of low-molecular-weight metabolites by (1)H nuclear magnetic resonance spectroscopy. Anal Biochem. 2002 May 15;304(2):220-30. http://www.ncbi.nlm.nih.gov/pubmed/12009699
[17]. https://www.caymanchem.com/pdfs/700460.pdf
[18]. http://usfiles.us.szc.pl/pliki/plik_1262018104.pdf
[19]. http://www.wiener-lab.com/wiener/catalogo/archivos/6322_creatinina_enzimatica_aa_liquida_pl.pdf
[20]. http://www.diag.pl/Badanie-Klirens-kreatyniny.85+M51588beb2ff.0.html
[21]. Fossati P., Prencipe L., 2010. Chromogenic System for Measuring Hydrogen Peroxide: The Enzymatic Uric Acid Assay. Clinical Chemistry May 2010 vol. 56 no. 5 865-866. http://www.clinchem.org/content/56/5/865.full
[22]. http://chemklin.sum.edu.pl/uploaded/kreatynina.pdf
[23]. Bugajska J., Berska J., Hodorowicz-Zaniewska D., Sztefko K., 2010. Walidacja metody oznaczania kwasów tłuszczowych frakcji fosfolipidów w surowicy krwi. diagnostyka laboratoryjna, Journal of Laboratory Diagnostics 2010 • Volume 46 • Number 2 • 125-130. http://diagnostykalaboratoryjna.eu/journal/DL_2_2010._str_125-130.pdf
[24]. Bezpośrednia metoda kolorymetryczna do oznaczania żelaza w surowicy krwi lub osoczu. Fer-color, Wiener-lab. http://www.wiener-lab.com/wiener/catalogo/archivos/12055_fer_color_aa_liquida_pl.pdf
[25]. Elmagirbi A., Sulistyarti H., Atikah, . Study of Ascorbic Acid as Iron (III) Reducing Agent for Spectrophotometric Iron Speciation. J. Pure App. Chem. Res., 2012, 1(1), 11-17. http://jpacr.ub.ac.id/index.php/jpacr/article/view/101/98
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje