
|

Zamknij X
|




Wstęp
Mitochondria to pół autonomi- czne organella komórek eukariotycznych, których główną funkcją jest produkcja ATP, niezbędnego do prawidłowego funkcjonowania komórek. Posiadają one własny genom, a także aparat biosyntezy białek. Mutacje w genomie mitochondrialnym (mt-DNA) lub genomie jądrowym (nDNA), kodującym białka specyficzne dla mitochondriów, są przyczyną powstawania chorób mitochondrialnych, zwanych mitochondriopatiami. Choroby mitochondrialn dotykają tkanek o największym zapotrzebowaniu na energię. Mutacje mt-DNA mogą służyć jako biomarkery tych chorób oraz jako czynniki predykcyjne przebiegu choroby. Mitochondrialna terapia genowa daje możliwość komplementacji zmutowanych mt-DNA oraz produktów jego ekspresji przyczyniając się do wyleczenia lub zmniejszenia nasilenia objawów chorobowych [1].
Budowa i funkcje mitochondrium
Mitochondria są otoczone dwoma błonami, zbudowanymi z dwuwarstwy lipidowej (Rys.1). Zewnętrzna błona mitochondrialna w większości zbudowana jest z fosfolipidów i białek. Zawiera ona białka transbłonowe, tzw. poryny, które tworzą hydrofilowe kanały umożliwiające dyfuzję cząsteczek o masie 5-10 kDa. Powierzchnia zewnętrznej błony mitochondrialnej jest gładka, w przeciwieństwie do błony wewnętrznej, która jest silnie pofałdowana w liczne grzebienie mitochondrialne. Liczba tych uwypukleń jest związana z nasileniem procesu fosforylacji oksydacyjnej. Skład wewnętrznej błony mitochondrialnej różni się od składu błony zewnętrznej, większą zawartością białek i obecnością fosfolipidokardiolipiny. We wnętrzu tej błony znajdują się białkowe czynniki sprzęgające F0F1, które posiadają aktywność syntetazy ATP. Białka te stanowią około 15% wszystkich białek wewnętrznej błony mitochondrialnej. Pomiędzy wewnętrzną i ze-wnętrzną błoną mitochondrialną znajduje się przestrzeń międzybłonowa [2].
Zewnętrzna błona mitochondrialna przepuszcza większość jonów i rozpuszczalnych w wodzie substancji o dostatecznie niskiej masie. Natomiast błona wewnętrzna jest przepuszczalna, na drodze osmozy, jedynie dla tlenu, dwutlenku węgla, amoniaku, wody oraz substancji hydrofobowych. Jest natomiast nieprzepuszczalna dla cukrów, większości aminokwasów, fosforanów nukleozydów, koenzymu A i jego estrów. Transport tych substancji przez wewnętrzną błonę mitochondrialną jest możliwy dzięki specyficznym substratowo białkom błonowym (translokazy lub permeazy), które w warunkach korzystnego gradientu stężeń działają na zasadzie transportu biernego lub transportu aktywnego. Przepuszczalność błon mitochondrialnych decyduje o transporcie aktywnych substancji do wnętrza mitochondriów [2].
Wnętrze mitochondrium wypełnia matriks, w którym znajdują się białka, głównie enzymy cyklu kwasów trójkarboksylowych, β-oksydacji kwasów tłuszczowych, katabolizmu aminokwasów oraz enzymy biorące udział w syntezie mt-DNA, mt-RNA i białek. W macierzy mitochondrialnej zlokalizowane są również mitchondrialne DNA, rybosomy, tRNA i rRNA [3].
Ludzki genom mitochondrialny zawiera 4-10 cząsteczek kolistego DNA o długości około 16 kpz. Mitochondrialny DNA zawiera 37 genów kodujących: 2 cząsteczki rRNA, 22 tRNA oraz 13 polipeptydów. Wszystkie polipeptydy kodowane przez mt-DNA są podjednostkami kompleksów enzymów systemu fosforylacji oksydacyjnej. Pozostałe białka mitochondrialne są kodowane w genomie jądrowym, syntetyzowane na cytoplazmatycznych rybosomach i importowane do odpowiedniego subkompartmentu mitochondriom. Geny mitochondrialne ludzi nie zawierają intronów i z wyjątkiem jednego systemu regulatorowego, sekwencje międzygenowe są nieobecne lub są ograniczone do kilku zasad. W każdej komórce występują setki kopii mtDNA (poliplazmia) i w prawidłowych komórkach są one identyczne (homoplazmia), gdyż pochodzą tylko od jednego z rodziców, są przekazywane wyłącznie w linii żeńskiej. Heteroplazmia powstaje na skutek mutacji. Wówczas w komórce występuje więcej niż jeden rodzaj mtDNA. W mitochondrialnym DNA znajduje się tzw. obszar hiperzmienny. Jest to niekodujący fragment genomu mitochondrialnego, który różni się między ludźmi i jest wykorzystywany do badań genetycznych [3].
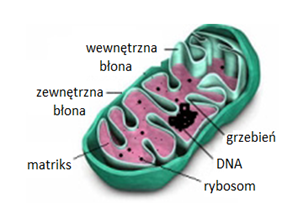
Rys.1. Schemat budowy mitochondrium [18].
Podstawową funkcją mitochondriów jest dostarczanie komórce energii w postaci ATP, produkowanego w procesie fosforylacji oksydacyjnej. Pozostałe funkcje mitochondriów związane są z metabolizmem nukleotydów, aminokwasów, lipidów, z procesem homeostazy jonowej oraz z udziałem tych organelli w procesie apoptozy. Lokalizacja mitochondriów w komórce nie jest stała. W wyniku ruchów cytoplazmy lub dzięki związaniu się z elementami cytoszkieletu, mitochondria mogą przemieszczać się w kierunku miejsca o zwiększonym zapotrzebowaniu na energię. Również liczba mitochondriów w komórkach nie jest stała, mieści się w zakresie od kilku do kilku tysięcy w komórce, i zależy od zapotrzebowania komórki na energię. Organy, takie jak wątroba, mózg, mięsień sercowy czy mięśnie szkieletowe, wykazujące dużą aktywność metaboliczną tlenu, zawierają znacznie większą ilość mitochondriów niż pozostałe, z tego też powodu najbardziej ze wszystkich organów są podatne na mutacje genomu mitochondrialnego [2].
Choroby mitochondrialne
Mutacje w genomie mitochondrialnym oraz jądrowym, który koduje około tysiąca białek mitochondrialnych, mogą być przyczyną zaburzeń prawidłowego funkcjonowania mitochondriów. Częstotliwość powstawania mutacji w obrębie mitochondrialnego DNA jest wielokrotnie większa niż w jądrowym DNA. Prawdopodobnie jest to związane z obecnością reaktywnych form tlenu (ROS) produkowanych przez mitochondrialny system oksydacyjnej fosforylacji oraz brakiem systemów naprawczych i ochronnego działania histonów. Mutacje rejonów kodujących w mt-DNA powodują zmiany w sekwencjach białek łańcucha oddechowego oraz mitochondrialnych rRNA i tRNA. Natomiast mutacje rejonu niekodującego mogą wpływać na zaburzenia w procesie replikacji i transkrypcji mt-DNA. Zwiększoną akumulację mutacji mitochondrialnego DNA obserwuje się w starzejących się tkankach oraz w wielu stanach chorobowych, takich jak schorzenia neurologiczne, metaboliczne, w stanach przednowotworowych oraz w niektórych nowotworach. Stan chorobowy zależy od ilościowego stosunku prawidłowych mt-DNA do zmutowanych, przy czym ujawnia się dopiero po przekroczeniu pewnej wartości progowej [4].
Choroby mitochondrialne są trudne do zdiagnozowania, a ich leczenie jest objawowe. Najczęściej dotyczą one tkanek o największym zapotrzebowaniu energetycznym, tkanki mięśniowej i nerwowej. Chorobom mitochondrialnym towarzyszą zaburzenia fosforylacji oksydacyjnej i kompleksów łańcucha oddechowego, brak aktywności niektórych enzymów oraz zmiana ultrastruktury mitochondriów. Choroby mitochondrialne mogą być powodowane przez zaburzenia jednogenowe oraz wieloczynnikowe [5].
Za występowanie zaburzeń jednogenowych odpowiada konkretna mutacja w obrębie pojedynczego genu, która jest odpowiedzialna za dysfunkcję łańcucha oddechowego. Mutacja ta stanowi czynnik wysokiego ryzyka rozwoju choroby. Do tego typu chorób mitochondrialnych, w których stwierdzono bezpośredni związek dysfunkcji mitochondriów z mutacjami mt-DNA można zaliczyć m.in.:
Zaburzenia wieloczynnikowe spowodowane są przez jednoczesny wpływ przyczyn genetycznych i środowiskowych. Mutacje w mt-DNA nie są wystarczające do wystąpienia choroby. Natomiast odpowiadają one za genetyczną skłonność do wystąpienia danego schorzenia, które ujawnia się, gdy dodatkowo zadziałają jeszcze określone czynniki środowiskowe i przekroczony zostanie próg podatności. Do tego typu chorób mitochondrialnych, w których mutacje w mt-DNA mogą być czynnikami ryzyka, można zaliczyć:
Cechą wspólną wymienionych chorób wieloczynnikowych jest ich związek z procesem starzenia, co może być wynikiem akumulacji mutacji w mt-DNA. Istnieje także możliwość, że choroby te mogą nie wynikać z uszkodzenia mitochondriów, ale ich rozwój prowadzi do takich zmian w funkcjonowaniu mitochondriów, że stają się one markerem choroby [5].
Mitochondrialna terapia genowa Mitochondrialna terapia genowa polega na naprawie zmutowanego mt-DNA lub produktów jego ekspresji. Można wyróżnić dwie strategie mitochondrialnej terapii genowej: pośrednią i bezpośrednią. Pośrednia mitochondrialna terapia genowa (zwana także ekspresją allotopową) występuje wówczas, gdy terapeutyczny gen, a w warunkach fizjologicznych składnik mt-DNA, jest przenoszony, z wykorzystaniem wirusowych lub nie wirusowych wektorów, do jądra komórkowego. W jądrze ulega on transkrypcji przez enzymy jądrowe, a następnie jest transporto-wany do wnętrza mitochondrium przez mitochondrialne peptydy sygnałowe, które uruchamiają fizjologiczny mechanizm importu białek (Rys.2). Pośrednia mitochondrialna terapia genowa posiada wiele trudnych do pokonania problemów, które mogą dyskwalifikować tę strategię jako potencjalne rozwiązanie terapeutyczne. Są to między innymi:
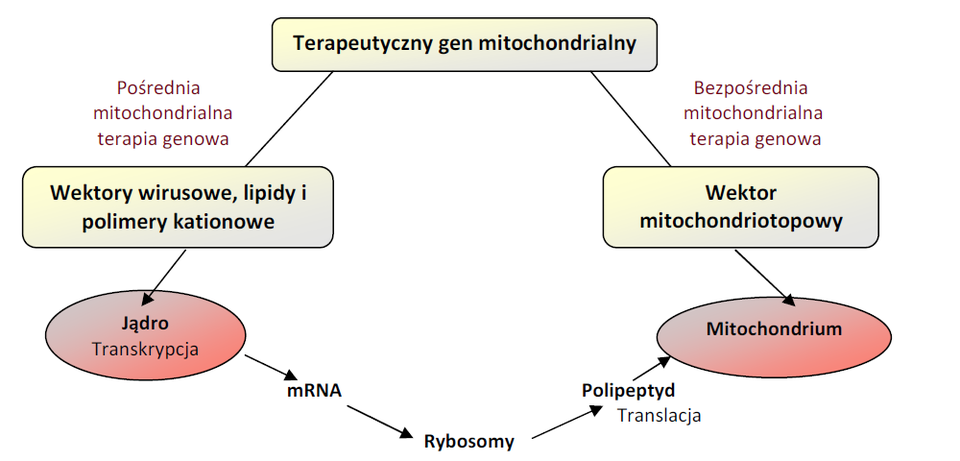
Rys.2. Schemat pośredniej i bezpośredniej strategii mitochondrialnej terapii genowej.
Bezpośrednia mitochondrialna terapia genowa umożliwia korygowanie mutacji bezpośrednio w mt-DNA (Rys.2). Istnieją dwie podstawowe strategie postępowania, stosowane w bezpośredniej mitochondrialnej terapii genowej, polegające na:
Pomimo tego, że obie strategie wykorzystują różne geny terapeutyczne, dają ten sam rezultat, polegający na zmniejszeniu objawów chorobowych na skutek zwiększenia się proporcji prawidłowych mt-DNA w stosunku do zmutowanych mt-DNA [6].
Systemy umożliwiające transport kwasów nukleinowych z cytoplazmy do mitochondriów
Istotnym zagadnieniem mitochondrialnej terapii genowej jest transport DNA do komórki, a następnie do wnętrza mitochondriów. Istotne jest, aby DNA uwalniane było w bezpośredniej bliskości mitochondrium, w kontakcie z zewnętrzną i/lub wewnętrzną błoną mitochondrialną, gdzie po uwolnieniu będzie mogło zostać przeniesione do macierzy mitochondrialnej [7].
Dotychczas do transportu kwasów nukleinowych do mitochondriów używano systemów transferu genów przez błonę komórkową na drodze endocytozy czy endocytozy receptorowej. Jednak zastosowanie tego systemu powodowało degradacje transportowanych biomolekuł w lizosomach lub endosomach. Obecnie prowadzone są badania nad zastosowaniem peptydów CCPs do transportu molekuł, takich jak DNA, białka czy peptydy, bezpośrednio do cytozolu. Peptydy CCPs są to krótkie, dodatnio naładowane sekwencje 10-30-aminokwasowe, transportowane przez błonę komórkową komórek ssaczych, co umożliwia ich wykorzystanie w terapii genowej [6,7]. Badane są również peptydy HIV-TAT i HSV-VP22. Są to strukturalnie podobne peptydy, zawierające mniej niż 20 aminokwasów w sekwencji, w tym znaczne ilości argininy i lizyny. Sekwencje te określa się jako PTD. Peptydy PTD, szczególnie TAT, są wydajnymi nośnikami peptydów, oligonukleotydów, białek i liposomów. Okazały się one również efektywnym nośnikiem peptydów do mitochondriów [8].
Systemy transportu makromolekuł do mitochondriów wykorzystują dwie zasadnicze cechy tych organelli: fizjologiczny import mitochondrialnych białek oraz wysoki potencjał błon mitochondrialnych. Wyższy niż w innych organellach wewnątrz komórki potencjał błon mitochondrialnych umożliwia gromadzenie się dodatnio naładowanych lipofilowych kationów w mitochondriach. Podstawowymi rodzajami transportu makromolekuł do mitochondriów są:
Możliwe jest także wprowadzenie pDNA do mitochondriów przy zastosowaniu metod fizycznych, takich jak: mikroiniekcja, „strzelby biolistyczne" oraz elektroporacja. Jednak zastosowanie tych metod jest ograniczone do izolowanych mitochondriów [6,9].
Białka transportowane do mitochondriów są syntetyzowane jako preproteiny na cytozolowych rybosomach. Zawierają one N-końcową mitochondrialną sekwencję liderową (MLS) o długości 3—60 aminokwasów i odpowiedniej strukturze pierwszorzędowej, w skład której wchodzi kilka aminokwasów hydrofobowych i kilka aminokwasów dodatnio naładowanych. Sekwencja ta jest rozpoznawana przez receptory na powierzchni zewnętrznej błony mitochondrialnej, co uruchamia mechanizm transportu białek. Receptory dostarczają preproteiny do kompleksu translokazy zewnętrznej błony mitochondrialnej (TOM), który transportuje części białek budujących zewnętrzną błonę mitochondrialna oraz białek występujących w przestrzeni międzybłonowej mitochondriów. W celu transportowania pozostałych białek, TOM współpracuje z innymi translokazami [6,10].
Translokazą odpowiedzialną za transport większości białek matriksu i białek budujących wewnętrzną błonę mitochondrialną jest kompleks TIM 23. Kompleks ten może rozpoznawać różnego rodzaju sekwencje kierujące, jednak w większości przypadków rozpoznaje on odpowiednią, obdarzoną ładunkiem dodatnim, N-terminalną sekwencje aminokwasową. Sekwencja ta jest następnie usuwana proteolitycznie przez odpowiednią matriksową peptydazę. Natomiast importowane białko jest wiązane i uwalniane przez białko mtHsp70. Kompleks TIM 23 korzysta z dwóch źródeł energii: potencjału błonowego i ATP w procesach transportu i wbu-dowywania białek w błonę mitochondrialną [6,10].
Mitochondrialne sekwencje liderowe (MLS) służą jako nośnik do transportu białek do wnętrza mitochondriów poprzez wytworzenie wiązania kowalencyjnego MLS - białko mitochondrialne lub MLS - białko niemitochondrialne. Prowadzone są badania nad łączeniem izolowanych mitochondriów i koniugatów mitochondrialnych peptydów liderowych (MLS) z kwasami nukleinowymi w postaci oligodeoksyrybonukleotydów, DNA lub PNA. Wykazano, że koniugaty te są zdolne do przechodzenia przez zewnętrzną i wewnętrzną błonę mitochondrialną z wykorzystaniem białek transbłonowych. Świadczy to o możliwości zastosowania mitochondrialnego importu białek z cytozolu do mitochondriów do transferu genów. Dzięki temu istnieje możliwość korekty zmutowanych mt-DNA w wyniku wprowadzenia dokładnej kopii genów prawidłowego mt-DNA [11].
Prowadzono także badania nad wykorzystaniem systemu PTD-MLS do transportu egzonukleazy III (Exo III) do matriksu mitochondrialnego. MLS połączono kowalencyjnie z N-końcem białka Exo III, zaś TAT z C-końcem tego białka, otrzymując białko fuzyjne MLS-ExoIII-TAT. Fuzja ta miała na celu stworzenie koniugatu, w którym występuje obok siebie TAT - molekularny nośnik przez błonę komórkową - oraz MLS - nośnik przez błony mitochondrialne. Zastosowanie systemu PTD-MLS umożliwia także transport innych bioaktywnych molekuł, takich jak DNA czy oligodeoksyrybonukleotydy, co może mieć zastosowanie w mitochondrialnej terapii genowej [12].
Zastosowanie endonukleaz restrykcyjnych, specyficznych dla komórek zmutowanych w obszarze mt-DNA, pozwoliło na selektywną degradacje zmutowanych mt-DNA. Zaobserwowano, że u pacjentów chorych na NARP mutacja T8399G generuje specyficzne miejsce restrykcyjne dla enzymu Pstl, które nie występuje w zdrowych mt-DNA. Skonstruowanie koniugatów DNA (gen Pstl) - MLS (mitochondrialną sekwencja liderowa) i ich transport do mitochondriów oraz ekspresja genów w ludzkich komórkach wywołała eliminację mutantów mt-DNA [13].
Transport pDNA przez błony mitochondrialne jest nie możliwy ze względu na wysoką barierę energetyczną oraz nukleotropizm. Prowadzone są badania nad utworzeniem koniugatów transportowanej molekuły z cząsteczką o tropizmie mitochondrialnym. Cząsteczki mitochondriotopowe, określane jako zdelokalizowane kationy (DLCs), są amfifilowe oraz zawierają sprzężony układ elektronów π, który umożliwia delokalizację ładunku dodatniego. Do tego typu związków należą m.in. rodamina 123, chlorek dekaliniowy oraz kationowe sole arylofosfoniowe. Utworzenie koniugatów mitochondriotopowych cząsteczek z DNA umożliwia ich przeniesienie przez błonę komórkową i transport w kierunku mitochondriów [6,14].
Do transportu koniugatów peptyd - kwas nukleinowy (PNA) do mitochondriów wykorzystano kation trifenylofosfoniowy (TPP). W celu uzyskania selektywnego hamowania repli-kacji zmutowanych DNA w stosunku do zdrowych, zastosowano 11-nukleotydowe sekwencje oligomerów komplementarnych do nici zmutowanych mitochondrialnych mRNA lub DNA. W przypadku heteroplazmii taka mitochondrialna terapia antysensowa mogłaby w sposób selektywny wspierać replikacje zdrowego mt-DNA w stosunku do zmutowanego mt-DNA. Umożliwiłoby to zwiększenie ilości zdrowego mt-DNA w komórce do poziomu zapobiegającego wystąpieniu symptomów klinicznych choroby mitochondrialnej [15].
DQAsomy są to pierwsze wektory specyficzne dla mitochondriów. Chlorek dekaliniowy wykazuje zarówno tropizm mitochondrialny, jak i tendencję do tworzenia miceli w warunkach wodnych. Ulega on szybkiej dyfuzji przez obie błony mitochondrialne i akumuluje się wewnątrz mitochondriów na skutek elektrostatycznego przyciągania przez mitochondria. DQAsomy wykazują podobieństwo do klasycznych kationowych liposomów. Podobnie jak kationowe liposomy, DQAsomy kompleksują DNA, chronią DNA przed enzymatyczną degradacją przez nukleazy, a ich cytotoksyczność jest porównywalna z wektorami kationowymi dostępnymi w handlu. Przeprowadzono liczne badania potwierdzające użyteczność DQAsomów i ich kompleksów z DNA jako nośników DNA w mitochondrialnej terapii genowej. Badania uwalniania się DNA z kompleksów DNA-DQAsomy na żywych komórkach wykazały endosomolityczną aktywność DQAsomów oraz ich skłonność do uwalniania DNA dopiero w kontakcie z błoną mitochondrialną bogatą w kardiolipinę. DQAsomy okazały się efektywnymi mitochondriotopowymi nośnikami koniugatów DNA-peptydowa sekwencja liderowa (MLS). Po uwolnieniu z DQAsomów, połączenia DNA-MLS wykorzystują natywny transport białek, aby dotrzeć do wnętrza mitochondriów [6,16].
Transport cytozolowych tRNA do dysfunkcyjnych mitochondriów - zmutowanych w obszarze genów kodujących mt-tRNA
Ponad 60% mutacji mitochondrialnego DNA dotyczy sekwencji kodujących mitochondrialne tRNA. Mutacje w genach mt-tRNA mogą powodować m.in. terminację transkrypcji, obniżenie stabilności cząsteczek mt-tRNA, brak rozpoznania mt-tRNA przez odpowiednią aminoacylo-tRNA syntetazę. Zaburzenia te skutkują nieprawidłowościami w przebiegu procesu translacji białek łańcucha oddechowego, co jest przyczyną niektórych chorób mitochondrialnych. Przeprowadzone badania wykazały, że istnieje możliwość importu in vitro tRNA do ludzkich mitochondriów, co umożliwia komplementację „wadliwych" mt-tRNA [6,17].
W zależności od typu organizmu i rodzaju RNA wyróżnia się cztery rodzaje transportu tych biomolekuł przez błony mitochondrialne:
Podsumowanie:
W ostatnich latach prowadzone są liczne badania nad ekspresją informacji genetycznej w mitochondriach oraz mutagenezą mitochondrialnego DNA. Mutacje w mt-DNA mogą indukować nieuleczalne choroby jednogenowe, jak również sprzyjać rozwojowi chorób wieloczynnikowych. Naprawa lub usunięcie zmutowanego mt-DNA lub jego komplementacja są jedynym sposobem na wyleczenie pacjentów lub zmniejszenie objawów chorobowych w przypadku pacjentów z chorobą wieloczynnikową. Mitochondrialna terapia genowa oparta na technologii mitochondriotopowych nośników DNA/RNA umożliwia kierowanie funkcjami mitochondriów i powinna przynieść wiele korzyści terapeutycznych. Wiąże się duże nadzieje z rozwojem mitochondrialnej terapii genowej. Jednakże dotychczasowe badania prowadzone są jedynie na izolowanych mitochondriach, a bardzo rzadko na modelach zwierzęcych. Dlatego możliwość zastosowania mitochondrialnej terapii genowej w badaniach klinicznych wymaga prowadzenia dalszych badań.
Słowa kluczowe: choroby mitochondrialne, mitochondrialny DNA, mitochondrialne tRNA, mitochondrialna terapia genowa, endogenne kompleksy translokacyjne, koniugaty DNA, DQAsomy.
Literatura:
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje