W skład proteoglikanów wchodzą białka i węglowodany, które to występują w przeważającej większości (80–90%). Ponadto, proreoglikany (PG) zawierają dużo estrowych grup siarczanowych [7].
Proteoglikany (PG) zaliczane są do glikoprotein. Ich główną cechą jest to, że zawierają duże ilości komponentu cukrowego (aż do 90%) , a także uczestniczą w zorganizowaniu międzykomórkowej substancji podstawowej tkanki łącznej. Biorą też udział w tworzeniu specyficznych receptorów powierzchniowych, które „wmontowane„ są w błony plazmatyczne. Fizjologiczne właściwości proteoglikanów wynikają z ich zdolności do wiązania kationów, łatwego tworzenia kompleksów z niektórymi białkami (np. lizozymem bądź kolagenem), a także z ich wielkiej hydrodynamicznej objętości molekularnej [1].
Ponadto, wykazano , że natywne proteoglikany występują w tkankach w postaci suprakompleksów tj. agregatów proteoglikanowych (PGA), które to powstają w wyniku niekowalencyjnej agregacji dużej liczby podjednostek proteoglikanowych (PGS) z wysokopolimerową nicią kwasu hialuronowego. Tak więc, elementarną jednostką strukturalną jest PGS o charakterystycznym składzie chemicznym i strukturze przestrzennej [1].
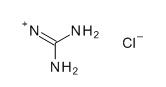
Metoda izolacji kompleksów proteoglikanowych z chrząstki za pomocą chlorku guanidyny [2], [1].
Za pomocą 4M chlorku guanidyny (GdnCl) możliwe jest całkowite wyekstrahowanie agregatów proteoglikanowych (PGA) , które są dość silnie związane z włóknami łącznotkankowymi. Ekstrakcja jest najefektywniejsza przy pH= 5,8. Jednocześnie, w środowisku 4 M roztworu GdnaCl dochodzi do odwracalnej dusocjacji agregatu PGA na elementy składowe tj.:
- PGS
- kwas hioluronowy oraz
- białka wiążące.
W celu zachowania integralności natywnej struktury PGA, do roztworu ekstrakcyjnego wprowadza się inhibitory grupowe proteaz, zaś oczyszczone cząsteczki PGA uzyskuje się ostatecznie na drodze ultrawirowania [2], [1].
Jako materiał wyjściowy stosuje się chrząstkę przegrody nosowej (lub tchawicy) bydlęcej (wołu), które pobiera się natychmiast po uboju i zamraża w termosie z lodem. Następnie, chrząstki oczyszcza się ze śluzówki oraz z ochrzęstnej, rozdrabnia się ją na małe kawałki oraz mieli w elektrycznej maszynce do mięsa. Wszystkie te zabiegi należy przeprowadzać w chłodni. Rozdrobnioną chrząstkę mrozi się w -20 - -40C [2], [1].
Zdjęcie: chlorek guanidyny, https://pl.vwr.com/app/catalog/Product?article_number=A4014.1000
Wykonanie:
Rozdrobnioną wcześniej chrząstkę należy rozmrozić, po czym 5g chrząstki należy zalać 100 ml 4 M roztworu GdnCl (tj. 4 M roztwór GdnCl w 50 mM buforze octanowym o ph=5,8, zawierającym inhibitory proteaz: 1 ml benzamidyny, 10 mM wersenianu sodowego i 100 mM kwasu e- aminokapronowego (EAK). Całość wymieszać dokładnie używając mieszadła szklanego (mieszać w chłodni przy małych obrotach przez 20 godzin [2], [1].
Po tym czasie opalizujący ekstrakt należy przesączyć na lejku z watą szklaną. Otrzymany w ten sposób klarowny roztwór należy rozdzielić na kilka woreczków dializacyjnych, po czym dializować go wobec 9 objętości 50 mM buforu octanowego z inhibitorami w chłodni. Otrzymany roztwór zawiera zasocjowane agregaty proteoglikanowe oraz część białęk nieproteoglikanowych ( w tym fragmenty kolagenu) [2].
Następnie, do połączonych dializatów należy dodać równą objętość etanolu zawierającego 1% CH3COOK, po czym należy odstawić w 4C na kilka godzin. Utworzony po odstaniu osad odwirować przy 10 000xg przez 10 minut. W tym czasie do odmierzonej objętości buforu octanowego (pH=5,8) dodać stałego CsCl do końcowego stężenia równego 1,69g/ml [2].
Osad PGA rozpuścić w 20 ml tego roztworu, po czym próbkę przenieść do cienkościennej plastikowej probówki wirówkowej i po dokładnym zrównoważeniu przenieść do głowicy kątowej 50 Ti (bądź podobnej ultrawirówki) i odwirować w temperaturze 20C przez 20h, przy prędkości 105 000 x g [2].
Po upływie czsu wirowania, dochodzi do grawitacyjnego rozwarstwienia mieszaniny białek próbki, a następnie do oddzielenia frakcji PGA . Agregaty proteoglikanowe z racji tego, że mają największą masę czasteczkową, gromadzą się w dolnej (1/3) słupa roztworu, z kolei kolagen mieści się w szczytowej warstwie. Frakcje dolne (zawierające PGA) można można w najprostszy sposób uzyskać odciągając za pomocą pipety 2/3 górnego słupa roztworu [2].
Frakcje zawierające PGA należy połączyć , po czym dializować wobec 50 mM buforu octanowego o pH=5.8 w chłodni przez kolejne 12 godzin. Na tym etapie należy oznaczyć zawartość kwasu uronowego i białka w 1 ml roztworu, z kolei pozostałość roztworu zadać 2 objętościami 98% roztworu etanolu zawierającego 1% CH3COOK i odstawić w chłodni na kilka godzin. Utworzony osad odwirować przy 10 000xg, następnie przemyć etanolem, po czym wysuszyć w eksykatorze próżniowym. Wysuszony osad rozpuścić w 10 ml buforu octanowego (pH+5.8), po czym zamrozić w temperaturze -30C. Osad ten stanowi materiał wyjściowy do preparatyki podjednostek proteoglikanowych PGS [2].
Izolowanie podjednostek proteoglikanowych w gradiencie chlorku cezu [3], [1].
Oczyszczone agregaty proteoglikanowe (PGA) uzyskane na drodze wirowania w warunkach , które sprzyjają asocjacji, można zdysocjować w warunkach sprzyjających dysocjacji na podjednostki proteoglikanowe PGS , które to następnie oddziela się od pozostałych składników przez ultrawirowanie w gradiencie chlorku cezu [3].
Wykonanie:
Roztwór PGA w 50 mM buforze octanowym o pH=5.8 , należy rozcieńczyć za pomocą równej objętości 8 M roztworu chlorku guanidyny. Końcowe stężenie 4M roztworu GdnCl powoduje całkowitą dysocjację agregatów PGA. Następnie, roztwór doprowadzić za pomocą stałego chlorku cezu do wyjściowego stężenia 1,50 g/ml. Następnie, cały roztwór rozlać do przeźroczystych probówek plastikowych , po czym zwirować przez 20 godzin w temperaturze 20C, przy 105 000xg [3].
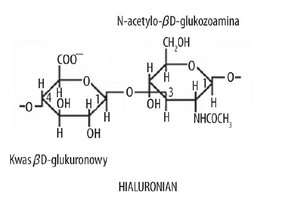
W czasie tego długotrwałego wirowania wytwarza się gradient chlorku cezu, elementy składowe zdysocjowanego agregatu ulegają rozdzieleniu w polu grawitacyjnym oraz gradiencie (zgodnie z ich masą cząsteczkową). Białka wiążące tworzą najwyższą warstwę gradientu, dzięki czemu łatwo jest je odciągnąć. Hialuronian zajmuje warstwę środkową, zaś w dolnej 1/3 słupa płynu gromadzą się wszystkie niejednorodne cząsteczki PGS [3].
Za pomocą pipety automatycznej odciągnąć 2/3 słupa płynu, pozostawiając PGS w dolnej reszcie. Wszystkie frakcje, które zawierają PGS połączyć i dializować wobec wody destylowanej w temperaturze 4C przez 12 godzin. Następnie, oznaczyć stężenie kwasu uronowego, heksazamin i białka w 1 ml roztworu. Pozostały roztwór zadać 2 objętościami etanolu, po czym próbkę odstawić do chłodni na 12 godzin. Na koniec osad odwirować, przemyć etanolem i wysuszyć w eksykatorze próżniowym [3].
Degradacja proteoglikanów przez proteazy [4], [1].
Proteazy, są enzymami, które odgrywają bardzo ważną rolę w każdej żywej komórce. U człwieka występuje ok. 500 różnych proteaz, które to kodowane są przez 2% wszystkich ludzkich genów. Proteazy odgrywają bardzo ważną rolę w wielu procesach,m.in. w trakcie zapłodnienia, trawienia, wzrostu, dojrzewania,a także starzenia się i śmierci organizmu [6].
Ponadto, regulują liczne procesy fizjologiczne przez kontrolę aktywacji, syntezy i degradacji białek. Proteazy pełnią także istotną funkcję w procesie replikacji i rozprzestrzenianiu się wirusów, bakterii oraz parazytów. Dodatkowo, enzymy te odpowiedzialne są za efektywną transmisję choroby wywoływanej przez wymienione patogeny. I to właśnie z tego powodu peptydazy stanowią istotny cel w przypadku leczenia wielu chorób. Nieprawidłowy poziom proteaz prowadzi do wielu nieprawidłowości fizjologicznych, czego konsekwencją jest choroba [6].
Podjednostka proteoglikanowa PGS składa się z nitkowatego rdzenia polipeptydowego oraz z przyłączonych do niego łańcuchów siarczanów chondroityny i siarczanu keratanu.
Proteazy mają zdolność rozrywania łańcuchów peptydowych w różnych miejscach, dzięki czemu powstają fragmenty o różnej masie cząsteczkowej. Dodanie do mieszaniny poreakcyjnej chlorku cetylopirydynowego (CPC) powoduje wypadanie z roztworu nierozpuszczalnych kompleksów nie rozłożonego PGS oraz powstałych peptydoglikanów. Z kolei, wprowadzenie kwasu trichlorooctowego powoduje rozpuszczenie peptydoglikanów (produktów enzymatycznego rozkładu) [4].
Wykonanie:
Przed przystąpieniem do przeprowadzenia doświadczenia, należy przygotować odpowiedni roztwór substratu tj. każdorazowo sporządzić świeży roztwór substratu o stężeniu 5 mg PGS w 1 ml odpowiedniego buforu:
- dla trypsyny: 50 mM bufor fosforanowy o pH= 7.8
- dla pepsyny: 50 mM bufor cytrynianowy o pH=3,6
- dla lizosomowej katepsyny D: 50 mM bufor octanowy pH= 5.0 [4], [1].
Roztwory proteaz w 1 ml odpowiedniego buforu:
1) 20 µg krystalicznej trypsyny
2) 50 µg krystalicznej pepsyny
3) 5 mg lizosomowej katepsyny D
Zdjęcie: http://www.phmd.pl/images/ryciny/20081223114758.jpg [5].
Do probówki wirówkowej typu Eppendorf wprowadzić 100 µl roztworu substratu PGS, próbkę preinkubować w temp. 37C przez 5 min. Następnie, do próbki dodać 50 µl próby enzymatycznej, dobrze wymieszać (próba B). Równolegle, nastawić próbę kontrolną (K) dodając w tym celu do substratu 50 µl wody. Próbkę inkubować w 37 przez 30 minut. Po upływie czasu inkubacji, działanie enzymatyczne należy pzrerwać dodając 50µl roztworu CPC (tj. 2,5% roztwór CPC), po czym próbkę odstawić w temperaturze pokojowej na 5 minut, zaś po upływie tego czasu dodać 100 µl 9% CCl3COOH i całośc dobrze wymieszać [4], [1].
Pozostawić w temperaturze pokojowej na kolejne 5 minut, Po tym czasie probówki przenieśc do łaźni lodowej (temp. 4-5C) na 30 minut. Po upływie tego czasu odwirować przy 10 000xg, w temperaturze 4C przez 10 minut [4].
Po wirowaniu, w klarownym supernatancie oznaczyć kwas uronowy metodą boranową stosując mikromodyfikację, tj. do probówek szklanych o poj. 5 ml oznaczonych: B (próba badana), K(kontrola), Wku(wzorzec kwasu uronowego: 250 µg kwasu glukuronowego w 1 ml H2O) i Wcs(wzorzec siarczanu chondroityny: 1000µg siarczanu chondroityny w 1ml H2O) wprowadzić 0,2 ml wody i 1,5 ml roztworu : stężony H2SO4- 0,025 M czteroboran sodu. Probówki wstawić do łaźni lodowej i ochłodzić, następnie na powierzchnię odczynnika boranowego nawarstwić po 50µl odpowiedniej próbki i trzymając nadal w łaźni wodnej zawartość probówek bardzo dokładnie wymieszać [4], [1].
Wszystkie próbki należy wstawić jednocześnie do wrzącej łaźni wodnej na 15 minut, po inkubacji próbki ochłodzić i dodać do nich po 50µl 0,125% roztworu karbazolu w etanolu bezwodnymi dobrze wymieszać, ponownie wstawić do łaźni wodnej na 6 minut. W tym czasie rozwija się zabarwienie prób. Po inkubacji próbki wyjąć z łaźni i odstawić do wystygnięcia. Na koniec dokonać odczytu absorbancji (dopiero po upływie 30 minut) wobec kontroli przy λ= 530 nm. W podobny sposób należy odczytać absorbancję wzorców. Powstała w próbkach barwa jest trwała przez 48h. Aktywnośc enzymu wyrazić liczbą µg kwasu uronowego oraz siarczanu chondroityny powstałych z rozkładu PGS w ciągu 1 minuty [4], [1].
Autor: Lidia Koperwas
Literatura:
[1]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s. 295-304.
[2]. Heinegard D., Hascall V.C, 1974. Agregation of cartilage proteoglycans, III. Characteristics of proteins isolated from trypsin digests of aggregates. J. Biol. Chem., 249: 4250-4256
[3]. Hascall V.C., Sajdera S.W, 1970. Phisical properties and polydispersity of proteoglycans from bovine nasal cartilage. J. Biol. Chem., 245: 4920-4926
[4]. Sapolsky A.J., Woessner J.F.jr., Howell D.S, 1975. A photometric assay for protease digestion of the N-acetylamino sugars. J. Biol. Chem., 217: 959-962.
[5].http://www.phmd.pl/images/ryciny/20081223114758.jpg
[6]. http://www.e-biotechnologia.pl/Artykuly/Proteazy/
[7]. http://nedo.gumed.edu.pl/wszpziu/skrypty/My%9Cliwski%20skrypt/tkanka%20%B3%B9czna.pdf
wstecz
Podziel się ze znajomymi









Recenzje