Kwasy nukleinowe, nukleotydy, nukleozydy i zasady azotowe mają zdolność selektywnego pochłaniania światła w nadfiolecie (UV). Maksimum przypada na 260 nm. Zjawisko absorpcji znalazło zastosowanie w analizie chemicznej. Właściwość ta wynika z obecności w kwasach nukleinowych układów purynowego lub pirymidynowego, które zawierają sprzężone wiązania podwójne. Na intensywność i charakter pochłaniania światła nie mają wpływu obecne reszty monocukrow, a także grupy fosforanowe, w związku z tym widma absorpcji zasad azotowych i odpowiadających im nukleotydów są bardzo podobne [7].Słowa kluczowe: absorbancja, spektrofotometria, oznaczanie kwasów nukleinowych, metoda Tsaneva i Markova, oznaczanie fosforu, zmodyfikowaną metodą Schmidta i Thannhausera, metamizol, metodą Horeckera , metodą Delory’ego
Absorbancja DNA nie jest addytywną sumą absorbancji wszystkich zasad azotowych wchodzących w jego skład, a to ze względu na fakt, że rzeczywista molowa absorpcja jest niższa o około 40% od wartości teoretycznie obliczonej na podstawie składu kwasu nukleinowego. Przedstawione zjawisko nosi nazwę efektu hipochromowego, jest ono związane z heliakalnym uporządkowaniem przestrzennym obu nici polinukleotydowych, opisanym strukturą drugorzędową i trzeciorzędową DNA [7].
Preparaty kwasów nukleinowych mogą być zanieczyszczone białkami, których maksimum pochłaniania światła przypada w paśmie długości równej 280 nm. Własności spektroskopowe makrocząsteczek pozwalają określić przybliżoną czystość preparatów kwasów nukleinowych. Obliczeń dokonuje się na podstawie wartości stosunku absorbancji przy 260 nm i 280 nm (A260/A280). Wolny od zanieczyszczeń dwuniciowy DNA (ds. DNA) ma wartość współczynnika A260 /A280/ równą 1,8, czysty RNA około 2, czyste białka poniżej 1 (około 0,5). Preparat DNA, którego wartość współczynnika A260 /A280 jest większa od wartości 1.8, może być zanieczyszczony RNA, gdy współczynnik A260/A280 ma wartość poniżej 1.8- można mniemać, że preparat zanieczyszczony jest białkami [7].
W przypadku pomiarów spektroskopowych zasadnicze znaczenie ma znajomość stężeń molowych kwasów nukleinowych. Stężenie kwasów nukleinowych można otrzymać z pomiaru absorpcji przy długości fali równej λ = 260 nm (jest to maksymalna absorpcja promieniowania nadfioletowego przez DNA)w przypadku, gdy znamy molowy współczynnik ekstynkcji ε:
C= A260/ ε • l
gdzie l – to długość drogi optycznej w kuwecie. Przyjmuje się, ze wartości współczynnika ekstynkcji
wynosi 6600 M-1 cm -1 [8].
Absorpcja kwasów nukleinowych zmierzona przy długości fali równej 230 nm odzwierciedla zanieczyszczenia pochodzące od węglowodorów, białek bądź fenolu. W przypadku czystych próbek wartość A260/A230 powinna wynosić 2,2. Z kolei, absorpcja zmierzona przy długości fali 325 nm może być wyznacznikiem wytrąceń w roztworze lub zanieczyszczeń pochodzących od samej kuwety [8].
Spektrofotometryczna metoda oznaczania kwasów nukleinowych według Tsaneva i Markova [2], [6].
W metodzie według Tsaneva i Markova, do rozdziału RNA i DNA w zasadowym hydrolizacie III, zaś do ekstrakcji DNA używa się HClO4,co pozwala przeprowadzić swobodnie w danych ekstraktach oznaczenia w nadfiolecie. Absorbancję otrzymanych ekstraktów RNA i DNA mierzy się przy dwóch długościach fali. Dla DNA jest to 268 i 284 nm, zaś RNA mierzy się przy 260 i 286 nm [2], [6].
Tabela: Postępowanie preparatywne – frakcjonowanie związków fosforowych według metody Tsaneva i Markova [2], [6].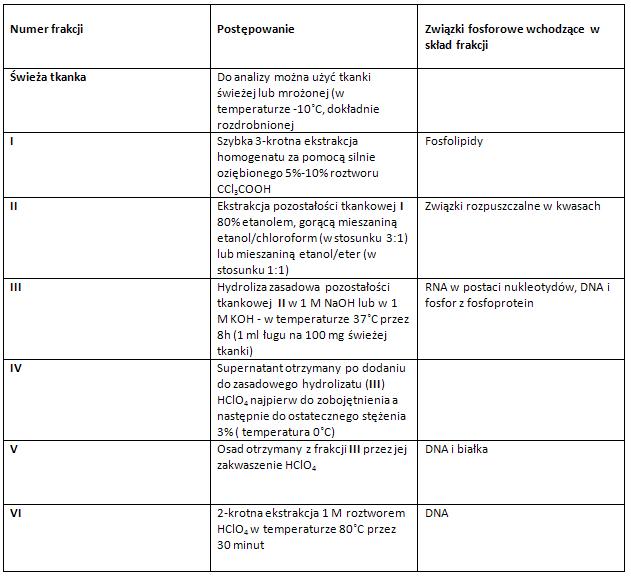 Oznaczanie zawartości związków fosforowych w tkance trzustki zmodyfikowaną metodą Schmidta i Thannhausera [3], [6].
Oznaczanie zawartości związków fosforowych w tkance trzustki zmodyfikowaną metodą Schmidta i Thannhausera [3], [6].Modyfikacja pierwotnej metody Schmidta i Thannhausera polega na zastosowaniu odlipidowania zgodnego z metodą Niemierki (1953), a także na zastosowaniu roztworów HClO4 do oddzielania DNA i związków rozpuszczalnych w kwasach [3], [6].
Jako materiał do przeprowadzenia oznaczenia używa się świeżej trzustki bydlęcej. Pierwszym etapem metody jest odlipidowanie tkanki. W tym celu należy sporządzić homogenat tkankowy z mieszaniną aceton/chloroform ( w stosunku 5:1) w homogenizatorze nożowym. Następnie, homogenat należy pobrać do probówki wirówkowej w ilości odpowiadającej 300 mg świeżej tkanki. Dalej przeprowadzić 3-krotną ekstrakcję za pomocą mieszaniny aceton/chloroform zmieszanych w stosunku 5:1 w temperaturze 0˚C, a dalej 3-krotną ekstrakcję mieszanina etanol/eter (3:1) w temperaturze 37˚C. Po każdej ekstrakcji próbkę odwirować w wirówce z chłodzeniem (1000 x g, 5 minut) [3], [6].
Supernatanty, które zawierają związki lipidowe należy połączyć i dopełnić jedną z mieszanin odlipidowujących do znanej objętości. Po tym etapie następuje ekstrakcja związków rozpuszczalnych w kwasach. W tym celu odlipidowaną tkankę ekstrahuje się w probówce wirówkowej 3-krotnie 0,2 M roztworem HClO4 (roztwór dodaje się porcjami po 4-5 ml)w temperaturze 0-4˚C przez 10 minut [3], [6].
Po każdej ekstrakcji próbkę należy zwirować w wirówce z chłodzeniem (ok. 2000 x g, 10 minut). Powstające supernatanty zbierać w cylindrze miarowym i dopełnić do 15 ml 0,2 M roztworem HClO4. Otrzymany osad związków nierozpuszczalnych w kwasach przemyć 2 razy zimnym 96% roztworem etanolu (dodawać porcjami po 4-5 ml), dalej przemyć 3-krotnie mieszaniną etanol/eter ( w stosunku 1:1), oraz 1 raz eterem. Osad wysuszyć na powietrzu, a następnie w eksykatorze próżniowym [3], [6].
Kolejnym etapem metody jest hydroliza związków nierozpuszczalnych w kwasach. W tym celu osad tych związków zalać 5 ml 1 M roztworu KOH, a po dokładnym wmieszaniu próbkę umieścić w cieplarce w temperaturze 37˚C, na 18 godzin. Dalej przeprowadzić rozdzielenie RNA i DNA- w tym celu otrzymany hydrolizat ochłodzić do 0˚C i zobojętnić w łaźni lodowej za pomocą 50% roztworu HClO4 ( wobec papierka wskaźnikowego), po czym do próbki dodać roztwór 50% HClO4 ( z takim wyliczeniem by końcowe stężenie HClO4 w hydrolizacie wynosiło 3%). Dokładnie wymieszać, a probówkę wirówkową umieścić w łaźni lodowej na 30 minut, aby uformował się osad. Po tym czasie próbkę odwirować ( ok. 1000 x g, 10 minut) w wirówce z chłodzeniem [3], [6].
Osad, który zawiera DNA po wirowaniu przemyć 3-krotnie za pomocą roztworu HClO4 (porcjami po 4-5 ml), po każdym przemyciu próbkę odwirować w wirówce z chłodzeniem (jak wyżej). Otrzymany po wirowaniu supernatant należy połączyć z cieczami z przemycia i dopełnić do objętości równej 20 ml. Osad przemyć 2 razy zimnym 96% roztworem etanolu (dodawanego porcjami po 4-5 ml), oraz 1 raz 96% roztworem etanol/eter (zmieszanych w stosunku 1:1) i 1 raz samym eterem [3], [6].
Ostatnim etapem metody jest oznaczenie zawartości fosforu metodą Horeckera (we wszystkich otrzymywanych w trakcie metody związkach tj. w całkowitym homogenacie, we frakcji lipidowej, w supernatancie- fosfor całkowity rozpuszczalny w kwasach, w supernatancie- fosfor RNA i fosfoprotein, oraz w osadzie- tj. DNA-P) [3], [6].
Zawartość fosforu w poszczególnych produktach wyliczyć w mg% świeżej tkanki- przeprowadzając bilans związków fosforowych. Ze względu na małą zawartość fosfoprotein w tkance trzustki fosfor supernatantu można (w przybliżeniu) przyjąć za fosfor RNA (RNA-P) [3], [6].
Oznaczanie zawartości fosforu metodą Horeckera i wsp. (1940) [1], [6].W trakcie badań nad enzymami stało się pożądane by opracować metodę, która pozwalałaby na określenie ilościowe nawet niewielkich ilości fosforu w białkach. Dostępne metody były niezadawalające ze względu na to, że wymagały albo przygotowania większej próby niż było to możliwe, albo dlatego , że nadmiar kwasu siarkowego, który był potrzebny do trawienia zakłócał obliczenia tj. określenie ilości fosforu w próbce [1], [6].
Kuttner i Cohen zgłaszali metodę, która umożliwiała określenie niewielkich ilości fosforu (1,7 μg), ale ilość kwasu siarkowego, który jest dozwolony w trakcie oznaczania fosforu był, z kolei niewystarczający do poprzedzającego trawienia . Berenblum and Chain byli w stanie określić mniej niż 1μg, ale ich metoda ma tę wadę, że wymaga ekstrakcji z niewielką ilością alkoholu izobutylowego [1], [6].
Poprzez modyfikację metody Fiske i Subbarowa i za pomocą spektrofotometru fotoelektrycznego opisane przez Hogness, Zscheile i Sidwell, 1 μg fosforu można określić z dokładnością do 3 procent. Aby zapewnić całkowite trawienie i uniknąć utraty fosforu, zwiększa się ilość używanego kwasu siarkowego. Końcowe stężenie kwasu siarkowego jest 2 M zamiast 0,5 M- tak jak jest określone w oryginalnej metodzie. Intensywność zabarwienia (kolor niebieski) kompleksu kwasu fosfomolibdenowego zwiększa się wraz z ogrzewaniem próbki- zgodnie z zaleceniami innych badaczy tj. Benedict i Theis. W temperaturze pokojowej kolor jest stabilny przez kilka godzin. Intensywność zabarwienia określa się spektrofotometrycznie i na podstawie określonego standardu oblicza się ilość fosforu [1], [6].
Metody kolorymetryczne powszechnie stosowane są do oznaczania fosforu nieorganicznego, który zależny jest od tworzenia heteropolikwasu „niebieskiego molibdenu”, z kwasu fosfomolibdenowego w wyniku stosowania różnego rodzaju środków redukujących, w tym np. metamizol (Guirgis i Habib, 1971) [5].
Metamizol jest solą sodową kwasu fenylo-dwumetylopirazolonometylaminometasulfonowego (C11H11N2O)?CH2SO3Na+H2O) (ryc. 1) – o masie cząsteczkowej 351. Jest on prawie białym, krystalicznym proszkiem, który łatwo rozpuszcza się w wodzie i alkoholu metylowym. Z kolei, jest trudno rozpuszczalny w spirytusie i całkowicie nierozpuszczalny w eterach.
Współczesna wiedza na temat metamizolu została przedstawiona w przeglądowej pracy Mészáros w 2001 r. [5], [9].
Metamizol (lub methampyron), niedrogi związek stosowany jako środek przeciwbólowy. W Polsce metamizol znany jest od wielu lat jako preparat o nazwie pyralgina i stosowany szeroko w leczeniu bólu, także tego pooperacyjnego. Pomimo, iż metamizol stosowany jest w praktyce lekarskiej od lat dwudziestych ubiegłego wieku, kiedy to został wprowadzony do leczenia w 1922 r., mechanizm działania tego związku został poznany bliżej dopiero w ostatnich latach [9].
W swoich doświadczeniach Guirgis i Habib (1971), zastosowali metamizol by sprawdzić czy będzie on przydatny przy ustalaniu ilości fosforu nieorganicznego w krwi i moczu, ponieważ wydaje się być dobrym związkiem redukującym [5].
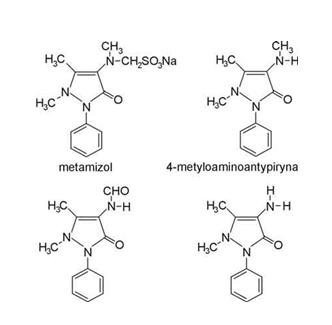 Rys. http://www.czytelniamedyczna.pl/174,metamizol-lek-ciagle-nowoczesny.html
Rys. http://www.czytelniamedyczna.pl/174,metamizol-lek-ciagle-nowoczesny.htmlW metodzie tej próbkę o zawartości fosforu od 3-10 μg należy zmineralizować (w probówkach) dodając 0,5 ml stężonego roztworu H2SO4 do pojawienia się białych dymów, po czym dodać 1-2 krople 72% roztworu HClO4. Po mineralizacji do probówki wprowadzić ok. 8 ml H2O, a po dokładnym wymieszaniu zawartości probówki dodać 0,5 ml 5% roztworu molibdenianiu (VI) amonu [6].Po ponownym wymieszaniu dodać 0,5 ml 0,2% roztworu eikonogenu (tj. sól sodowa kwasu 1,2,3-aminonaftolosulfonowego) lub amidolu (tj. chlorowodorku 2,4-diaminofenolu) z dodatkiem 12 g Na2S2O5 i 1,2 g Na2SO3). Całość dopełnić wodą do 10 ml. W podobny sposób należy wykonać próbę kontrolną [6].
Przygotowane próbki umieścić w łaźni wodnej na 15 minut, a następnie po ochłodzeniu należy oznaczyć absorbancję przy długości fali λ=720 nm. Aby odczytać zawartość fosforu w badanych próbkach należy sporządzić krzywą kalibracyjną w zakresie 1-15 μg fosforu (P) [6].
W metodzie tej intensywność zabarwienia mieszaniny niższych tlenków molibdenu zwiększa się wyniku ogrzewania analizowanych próbek we wrzącej łaźni wodnej przez 15 minut [6].
Oznaczanie zawartości fosforu nieorganicznego pochodzącego z fosfoprotein metodą Delory’ego [4], [6].
Opisana poniżej metoda jest przystosowana do oznaczania fosforanów nieorganicznych w obecności substancji przeszkadzających w redukcji kompleksu fosfomolibdenowego. Zasada metody opiera się na wytraceniu nieorganicznych fosforanów w postaci Ca3(PO4)2, osadzonego na MgCO3 [4], [6].
Roztwór, który zawiera nieorganiczny fosforan- pochodzący z fosfoprotein, należy odpipetować do skalowanych probówek ( o objętości 15 ml). Następnie, zawartość probówki zobojętnić ( w obecności fenoloftaleiny), dodając kroplami roztwór stężonego NH3, a po zobojętnieniu dodać dodatkowo 0,2 ml tego odczynnika. Do probówki wprowadzić 1 ml 2,5% roztworu CaCl2 oraz 1 ml 0,5% wodnej zawiesiny MgCO3. Zawartość probówki należy dokładnie wymieszać, po czym zostawić na 30 minut. Po tym czasie próbkę ponownie wymieszać i odwirować . Otrzymany po wirowaniu supernatant ostrożnie zlać (odrzucić), a ścianki probówki (wewnątrz) osuszyć bibułą [4], [6].
Otrzymany osad przemyć za pomocą 5 ml 2% roztworu NH3, próbkę odwirować, supernatant zlać a ścianki probówki osuszyć bibuła (jak wyżej). Dalej, osad rozpuścić w 1,1 ml 60% HClO4. Dodać ok. 10 ml H2O, 1 ml 5% roztworu molibdenianu (VI) amonu, 0,5 ml roztworu eikonogenu (tj. sól sodowa kwasu 1,2,3-aminonaftolosulfonowego: 2% roztwór eikonogenu z dodatkiem 12 g Na2S2O5 i 1,2 g Na2SO3) i uzupełnić wodą do objętości równej 15 ml. PO inkubacji próbki przez 10 minut, oznaczyć absorbancję roztworu w fotokolorymetrze przy λ= 720 nm. Zawartość roztworu należy obliczyć przez porównanie z absorbancją roztworów wzorcowych[6], [4].
Autor: Lidia KoperwasLiteratura:[1]. Horecker B.L., Ma T.S., Haas E, 1940. Note on the determination of microquantities phosphorus, 15 June 1940. J.Biol. Chem., 36: 775-776
[2]. Tsanev R., Markov G.G., 1960: Substances interfering with spectrophotometric estimation of nucleic acids and their elimination by the two-wavelength method. Biochim. Biophys, Acta, 42: 442-452.
[3].Schmidt G., Thannhauser S.J, 1945. J. Biol. Chem., 161: 83-99; Walter Z., 1970: Praca habilitacyjna, UŁ.
[4]. Delory C., Charles P., Ledoux L., 1938. A note on the determination of phosphate in the presence of interfering substances. Biochem. J., 32: 1161-1162.
[5]. Guirgis F.K., Habib Y.A., 1971.Use of a New Reducing Agent, Metamizol, in Determining Inorganic Phosphorus in Blood and Urine. Clinical Chemistry , Vol. 17, No. 2, 1971. S. 78-81
[6]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s. 351-359
[7]. [http://biochigen.slam.katowice.pl/praktikum/013.pdf]
[8].http://www.pg.gda.pl/chem/Katedry/Leki/Dydaktyka/Kultury/lab/Porownanie_ilosci_i_%20jakosci_DNA_wyizolowanego_z_komorki_roslinnej_i_zwierzecej.pdf
[9]. http://www.czytelniamedyczna.pl/174,metamizol-lek-ciagle-nowoczesny.html]










Recenzje