 Źródłem nowych leków są związki chemiczne znajdujące się w roślinach, organizmach zwierząt, lecz największą rolę w procesie tworzenia leku ma synteza nowych związków chemicznych w laboratoriach. Tam właśnie przeprowadza się pierwsze badania i odkrywa nowe ugrupowania chemiczne dające związek o istotnym działaniu biologicznym, który można zastosować w leczeniu lub zapobieganiu chorobom. Substancję modyfikuje się poprzez podstawienie do niej różnych grup chemicznych, uzyskując kilka wariantów o lepszych cechach w porównaniu z lekiem macierzystym.
Źródłem nowych leków są związki chemiczne znajdujące się w roślinach, organizmach zwierząt, lecz największą rolę w procesie tworzenia leku ma synteza nowych związków chemicznych w laboratoriach. Tam właśnie przeprowadza się pierwsze badania i odkrywa nowe ugrupowania chemiczne dające związek o istotnym działaniu biologicznym, który można zastosować w leczeniu lub zapobieganiu chorobom. Substancję modyfikuje się poprzez podstawienie do niej różnych grup chemicznych, uzyskując kilka wariantów o lepszych cechach w porównaniu z lekiem macierzystym.
Niekiedy jednak ukryte właściwości odkrywane są przypadkowo. Na przykład, w czasie prac nad lekiem stosowanym w schizofrenii odkryto przypadkowo pierwszy lek przeciwdepresyjny, natomiast pierwszy lek, który zastosowano w leczeniu schizofrenii został wyizolowany podczas prac nad lekiem przeciwalergicznym. Obecnie możliwe jest przewidzenie właściwości farmakologicznych nowego związku chemicznego zaplanowanego do syntezy.
Proces badawczo-rozwojowy nowego produktu leczniczego przebiega pod kontrolą nadzoru farmaceutycznego, za zgodą Lokalnej Komisji Bioetycznej. Składa się z następujących części, które muszą być opracowane i szczegółowo zaplanowane na długo przed właściwym rozpoczęciem badania:
• badania podstawowe (laboratoryjne)
• badania przedkliniczne
• badania kliniczne fazy I
• badania kliniczne fazy II
• badania kliniczne fazy III
Badania podstawowe- laboratoryjne są pierwszym etapem prac nad stworzeniem nowego leku. Ich celem jest synteza nowych związków chemicznych, poszukiwanie i izolacja substancji naturalnych (głównie z roślin), które mają potencjalne działanie lecznicze. Badania te opracowywane są w obszarze mechanizmów działania leku oraz mechanizmów odpowiedzialnych za powstawanie procesu chorobowego. Po wstępnej selekcji wyodrębnione substancje są poddawane badaniom przedklinicznym.
Pierwszy etap właściwych prac nad substancją, która ma znaleźć zastosowanie w medycynie, nazywany jest przedkliniczną fazą badań. W tej części testów związek musi przejść próby na komórkach in vitro oraz in vivo, podczas których ocenia się jego działanie. Następnie, jeśli wyniki są zadowalające lek może zostać podany zwierzętom doświadczalnym. Zwierzęta wykorzystywane są w badaniach medycznych jedynie, kiedy jest to bezwzględnie konieczne i nieuniknione, w sytuacjach, gdy nie istnieją odpowiednie alternatywne rozwiązania.
Badania in vivoBadania in vivo polegają na operacyjnym dojściu do danego narządu poddawanemu leczeniu i połączeniu go z przyrządami rejestrującymi jego czynność oraz sprawdzeniu działania różnych stężeń testowanej substancji na organizm. Badania operacyjne wykonuje się w znieczuleniu ogólnym, przestrzegając zasad etycznych. Jeśli istnieje taka możliwość, stosuje się nieoperacyjne badania narządów. W ostatnich latach zwiększyła się liczba metod badawczych nieoperacyjnych. Polegają one głównie na umocowaniu na różnych częściach ciała, w pobliżu badanych narządów urządzeń przekazujących do rejestratorów impulsy będące wynikiem czynności organu. Często jednak konieczne jest stosowanie obydwu rodzajów badań.
Badania in vitroBadanie in vitro polega na badaniu wpływu związku na wyizolowane z organizmu zwierzęcego narządy (serce, tchawicę, żołądek, jelita), tkanki lub pojedyncze komórki. Wyizolowaną próbkę umieszcza się w naczyniu doświadczalnym, w którym jest on omywany krwią lub płynem o składzie jonowym, kwasowości i temperaturze zbliżonej do warunków panujących w zdrowym organizmie zwierzęcym. W płynach ustrojowych (krew, mocz, płyn mózgowo-rdzeniowy) oraz w tkankach bada się wpływ nowego związku na normalne składniki ustrojowe, na zawartość białek, węglowodanów, tłuszczów, a także na aktywność enzymów.
Badania na zwierzętach są podstawowym i obecnie najlepszym sposobem oceny wpływu różnych substancji na organizm człowieka, pozwalają przewidzieć skutki ich oddziaływania, wyjaśnić przyczyny powstałej odpowiedzi i zachowania organizmu. Umożliwiają sprawdzenie działania np. kosmetyków, składników zawartych w badanym pokarmie czy na przykład wpływ środowiska na organizm.
Do doświadczeń używa się wyłącznie zwierząt nabytych legalnie, z hodowli prowadzonej według zasad zgodnych z obowiązującym prawem, których wykaz posiada Krajowa Komisja Etyczna ds. Doświadczeń na Zwierzętach. Niedopuszczalne są doświadczenia powodujące ból u zwierząt. W celu jego zniesienia stosuje się odpowiednie leki znieczulające i przeciwbólowe. Jeśli po zakończeniu doświadczenia zwierzę nie wymaga wybudzenia, powinno być ono zgładzone przed wybudzeniem z narkozy. Jeśli badania wymagają wybudzenia zwierzęcia, należy zapewnić mu pełną opiekę pooperacyjną, stosowaną w klinikach weterynaryjnych ( zniesienie niewygody i bólu).
Doświadczenia na zwierzętach mogą prowadzić wyłącznie osoby o odpowiednich kwalifikacjach, które posiadają indywidualne, numerowane zezwolenia uzyskane od kierownika jednostki doświadczalnej. Współuczestniczyć mogą osoby odpowiednio przygotowane, które również uzyskały zgodę kierownika jednostki doświadczalnej i Lokalnej Komisji Etycznej ds. Doświadczeń na Zwierzętach.
Pierwszym krokiem do prowadzenia badań jest ich zaplanowanie i przedłożenie projektu i wniosku do Komisji Etycznej Badań na Zwierzętach. Na skierowany wniosek, zgodę wydaje Lokalna Komisja Etyczna ds. Doświadczeń na Zwierzętach, która działa na podstawie Ustawy o doświadczeniach na zwierzętach z dnia 21.01.2005 r. i podlega Krajowej Komisji Etycznej ds. Doświadczeń na Zwierzętach. Badania można prowadzić jedynie w specjalnie przystosowanych do tego celu ośrodkach, zgodnie z art. 16 ust. 1 wyżej wymienionej Ustawy, których wykaz aktualizuje Ministerstwo Nauki i Szkolnictwa Wyższego.
Podawanie produktu różnym gatunkom zwierząt pomaga w przybliżonej ocenie cech nowej substancji. Badania farmakologiczne są prowadzone, co najmniej na trzech gatunkach zwierząt laboratoryjnych: dwóch niższych i jednym wyższym. Spośród zwierząt niższych są to najczęściej myszy, szczury, świnki morskie, chomiki, hodowane w ściśle określonych warunkach atmosferycznych, i odpowiednio karmione. Ze zwierząt wyższych do badań używa się królików, kotów lub psów. Jeśli istnieją szczególne przesłanki, efekty farmakologiczne nowych leków bada się także na zwierzętach naczelnych (małpy).
Nierzadko zdarza się, że oceniana substancja w organizmie zwierząt, zwłaszcza gryzoni, musi być zwielokrotniona by osiągnąć taki efekt, jaki może wywołać kilkukrotnie mniejsza dawka tej samej badanej substancji u człowieka.
Badania rozpoczyna się od określenia dawki śmiertelnej związku dla połowy z grupy badanych zwierząt, czyli od ustalenia tzw. DLM (dosis letalis). Dawkę tę bada się na trzech gatunkach zwierząt, dwóch niższych - np. myszy, szczury i jednego wyższego -króliki, koty, psy itp.
DLM i DE50Oprócz DLM w czasie badania określa się również DE50 (dosis effectiva) jest to dawka wywołująca efekt farmakologiczny u połowy z grupy badanych zwierząt. DE50 oblicza się za pomocą prostych metod statystycznych, po podaniu różnych dawek grupom liczącym od 5 do 10 zwierząt. Im wyższy wskaźnik leczniczy tym lek jest bardziej bezpieczny. Jeśli wskaźnik jest mniejszy od 1, związek nie może zostać zastosowany w lecznictwie ze względu na zbyt dużą toksyczność.
Przy użyciu metod chemicznych bada się stężenie badanego związku, po podaniu jego różnych dawek, w płynach ustrojowych, tkankach i na tej podstawie określa przypuszczalne dawki lecznicze. Stężenie badanego związku w moczu pozwala na określenie prędkości jego wydalania przez nerki. Zbadanie stężenia związku w tkankach daje informacje o wielkości jego dystrybucji do poszczególnych tkanek, narządów oraz ich części. Jeśli istnieje możliwość wywołania choroby doświadczalnej (np. zakażenia bakteryjnego, stanu zapalnego, nadciśnienia tętniczego, niedokrwienia serca, cukrzycy) bada się wpływ związku na przebieg choroby wywołanej u zwierząt, a także występowanie działań ubocznych. Oznaczane jest także mutagenne działanie badanej substancji.
Szacuje się również bezpieczeństwo stosowania badanej substancji u ludzi poprzez określenie: toksyczności ostrej i toksyczności przedłużonej. Badania nad toksycznością związku prowadzi się, aby wykryć jego ewentualną szkodliwość i ustalić wypływające stąd przeciwwskazania.
W tym celu związek podaje się zwierzętom laboratoryjnym przez okres od 6 do 12 miesięcy i bada się stan czynnościowy narządów- przede wszystkim szpiku kostnego, wątroby, nerek, układu sercowo-naczyniowego, pokarmowego i ośrodkowego układu nerwowego. Stosuje się metody fizjologiczne, biochemiczne i morfologiczne. Wyniki tych badań określają stopień szkodliwości, która może wystąpić po długotrwałym podawaniu związku. Podawanie związku miejscowo na skórę i błony śluzowe, pomaga określić jego ewentualne właściwości alergiczne.
Każdy nowy związek poddawany jest również badaniom embriotoksycznym, teratogennym i karcinogennym. W tym celu podaje się go co najmniej trzem gatunkom zwierząt w okresie ciąży i obserwuje czy powoduje on zmiany toksyczne u płodu lub deformacje jego narządów (teratogenność – potworniakowatość). Po kilku lub kilkunastomiesięcznym okresie podawania obserwuje się też, czy nie wywołuje on działania rakotwórczego (karcinogennego). Związki, które przechodzą te testy negatywnie zostają odrzucone, jako potencjalne leki.
Wykonuje się setki badań na dużej liczbie zwierząt. Każda technika wymaga użycia kilku grup liczących od 6 do10 zwierząt. Poszczególnym grupom podaje się różne dawki badanych związków. Każdą grupę po podaniu badanej substancji obserwuje się i porównuje się z grupą zwierząt kontrolnych, którym podaje się roztwór służący do jego rozpuszczenia, lub u których wykonuje się zabiegi przygotowujące do wprowadzenia związku do organizmu. Dzięki tym porównaniom uzyskuje się wyniki obiektywne, które nadają się do oceny statystycznej. Do oceny wyników dobiera się odpowiednią dla danego modelu metodę analizy statystycznej. Złe dobranie może powodować zafałszowanie oceny uzyskanych danych. Wyniki dają informacje, na jakie narządy i tkanki nowy związek wywiera działanie lecznicze. Z informacji tych można wnioskować, jakie działanie lecznicze wystąpi u ludzi.
Badania przedkliniczne nawet, gdy wypadną pozytywnie nie dają gwarancji, co do pomyślnego zakończenia badań klinicznych. Mogą jedynie mają pomóc w początkowym ustaleniu działania badanych substancji i cząsteczek określonego związku, które w przyszłości mogą trafić do dalszych etapów badań klinicznych i stać się przydatnymi specyfikami do walki z chorobą.
Nie istnieje model zwierzęcy, który umożliwiłby zastąpienie eksperymentalnego podawania nowego produktu leczniczego ludziom. Zwierzęta często reagują na substancje chemiczne zupełnie inaczej.
Bywa, że wynik badań, który u zwierząt wypada korzystnie nie jest bezpieczny i korzystny u ludzi. Jednym z badań klinicznych, które po pozytywnym teście na zwierzętach przeszło do fazy badań na ludziach było badanie leku Fialurydyny. Grupie piętnastu pacjentów podawano lek, który miał być stosowany w terapii przewlekłych zapaleń wątroby typu B18. Po 13 tygodniach badań u jednego z badanych stwierdzono kwasicę mleczanową i postępującą niewydolność wątroby. Badania przerwano, a u 7 kolejnych pacjentów stwierdzono objawy ciężkiej hepatotoksyczności, postępującej kwasicy mleczanowej, narastanie żółtaczki i pogarszanie się czynności wątroby, nawet po odstawienia leku. W konsekwencji 5 pacjentów zmarło, dwóch przeżyło dzięki przeszczepowi wątroby. U innych pacjentów stwierdzono zapalenie trzustki, neuropatie i miopatie. Lek wycofano z badań klinicznych.
Może to wynikać z różnic gatunkowych np. mniejszej odporności, wrażliwości. Dla przykładu organizm świni jest najbardziej zbliżony pod względem fizjologii do organizmu człowieka, natomiast najlepszym modelem zwierzęcym dla oceny skuteczności proponowanego leku przed zastosowaniem u ludzi w badaniu in vivo jest szympans. Jednak w praktyce nie wszystkie badania można wykonywać na tych zwierzętach.
Czasem substancja korzystna dla ludzi okazuje się bardzo niebezpieczna dla zwierząt np. paracetamol stosowany powszechnie u ludzi może wywołać zaburzenia lub nawet obumarcie ciąży psa, jest również wysoce toksyczny dla kotów, nawet w niewielkiej dawce.
Szacuje się, że tylko 5 na 5000 substancji leczniczych, nad którymi rozpoczyna się prace, pomyślnie przechodzi tę fazę badań.
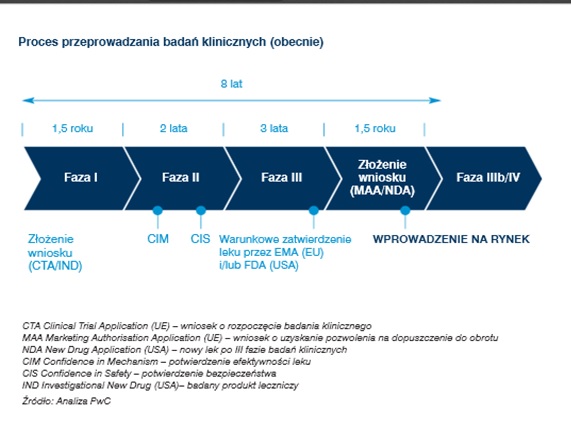
Po pomyślnym zakończeniu etapu badań przedklinicznych rozpoczynają się badania kliniczne, składające się z następujących faz I, II, III i IV. Każda z nich musi być zakończona pozytywnie, aby można było rozpocząć następny etap.
http://infarma.pl/fileadmin/badania_kliniczne_raport/Badania%20kliniczne%20w%20Polsce%202010.pdf
W trakcie I fazy badań wstępnie ocenia się bezpieczeństwo stosowania testowanego środka. Jest pierwszym kontaktem organizmu człowieka z nowym lekiem. Przeprowadzana jest zwykle na małej (zwykle od 10 do 100) grupie zdrowych ochotników. Badane jest jego wchłanianie z przewodu pokarmowego, poziom jaki osiąga we krwi i tkankach, wydalanie, toksyczność oraz interakcje z pożywieniem i powszechnie stosowanymi lekami. Wyniki tej części prac nad nowym lekiem pozwalają wstępnie określić jego dawkowanie poprzez badanie zależności pomiędzy dawką leku oraz odpowiedzią kliniczną.
W I fazie badań klinicznych bierze zazwyczaj udział kilkudziesięciu zdrowych ochotników jednakże w przypadku leków o potencjalnym działaniu toksycznym badaną próbę stanowią jedynie osoby chore. Ta faza trwa zwykle krótko, niesie ze sobą najwięcej ryzyka. Nowy lek podawany jest przez kilka dni, w dawce pojedynczej (w kilku stężeniach) oraz w dawkach wielokrotnych w różnych odstępach czasowych. Z udziału w tej fazie badań nie można odnieść bezpośrednich korzyści zdrowotnych.
Wzięcie udziału w tej fazie badań nad nowym lekiem może wymagać spędzenia pewnego okresu czasu w specjalnie przystosowanych do tego celu ośrodkach badawczych, należących do firm farmaceutycznych lub instytucji naukowych, w celu ścisłego monitorowania poziomu leku w organizmie. Przez cały czas trwania tej fazy uczestnicy są bardzo dokładnie badani i obserwowani. W przypadku niektórych leków, na przykład cytostatyków przeznaczonych do leczenia nowotworów, ta faza badań jest połączona z II fazą, ponieważ nie można narażać zdrowych ochotników na działanie silnie toksycznych związków.
Według danych amerykańskich na 100 leków poddawanych badaniom fazy I do dalszych badań przechodzi około 70.
Faza II badań klinicznychTa faza badań trwa zwykle przynajmniej 6 miesięcy. Udział w niej wiąże się prawie zawsze z koniecznością regularnych wizyt w klinice w celu monitorowania działania leku. Celem II fazy badań klinicznych jest stwierdzenie czy nowy lek działa u określonej grupy chorych i czy jest bezpieczny. Podczas tej części prac ustala się także związek pomiędzy dawką, a efektem działania preparatu, co pozwala na ostateczne ustalenie dawki stosowanej w dalszej fazie badań oraz dalszą ocenę skuteczności i bezpieczeństwa preparatu. Oceniane są szczegółowe parametry dotyczące wchłaniania, metabolizmu i wydalania leku w zależności od płci i wieku. Badania II fazy obejmują kilkuset ochotników (na ogół od 50 do 500) - pacjentów cierpiących na daną chorobę.
W II fazie testów prowadzi się pierwsze badania porównawcze działania nowego preparatu i placebo. Aby obiektywnie ocenić działanie nowego leku, badania te prowadzi się najczęściej metodą podwójnie ślepej próby- ani pacjent, ani badacz nie wiedzą, jaka substancja jest podawana choremu. Robi się tak, dlatego, by oczekiwania ani pacjenta, ani lekarza nie mogły wpłynąć na wyniki badań. Lekarz, który wie, czym leczony jest pacjent może bezwiednie inaczej postępować z pacjentem, natomiast pacjent znający swój sposób leczenia mógłby inaczej opisywać swoje ewentualne dolegliwości. Od dawna już wiadomo, iż pacjent przekonany, iż będzie dostawać nowy, bardzo skuteczny, trudno dostępny, drogi lek czuł się po nim znacznie lepiej, choć podawano mu tylko substancję obojętną – jest to tak zwany efekt placebo.
Uczestnicy tej fazy badań dzieleni są zwykle na dwie grupy: badaną i kontrolną. Znajdujący się w grupie badanej otrzymują testowany lek, grupa kontrolna- tak zwane placebo- nie działający, obojętny preparat, nie dający się odróżnić na podstawie wyglądu, smaku czy zapachu od badanego leku. Ze względów etycznych, coraz rzadziej używa się klasycznego placebo. Grupa kontrolna obecnie otrzymuje zwykle terapię uważaną aktualnie za standardowy sposób leczenia danego schorzenia.
„Przydział” do grupy odbywa się losowo, zwykle dokonuje go komputer (mówi się wtedy, że badania są randomizowane). Pozwala to, by osoby przydzielone do poszczególnych grup były do siebie podobne (były w podobnym wieku, stanie zdrowia, zawierały podobny odsetek kobiet), dzięki czemu możliwe będzie dokonanie porównania wyników leczenia. Osoba decydująca się na udział w badaniach klinicznych leku nie ma wpływu na to, do której grupy zostanie przydzielona. Nie ma na to wpływu także lekarz leczący. Informację o tym, do której grupy należał pacjent lekarz otrzymuje na ogół po zakończeniu badań.
Randomizacja, czyli losowy przydział do grup, często budził protesty. Pojawiały się głosy, iż pacjenci z zaawansowanym stadium danej choroby, nie mający już nowych opcji terapeutycznych, nie mogą czekać na zakończenie badań klinicznych, ryzykować przyjmowania placebo. Woleliby przyjmować mało znaną substancję badaną, która może, choć przecież nie musi, okazać się dla nich szansą na dłuższe życie. Te protesty głównie ze strony organizacji zrzeszających osoby żyjące z HIV spowodowały, iż od roku 1989 wprowadzono w USA sposób postępowania nazwany „ścieżką równoległą” (ang. parallel track) lub „rozszerzonym dostępem” (ang. expanded access). Polega na tym, iż leki, które pomyślnie przeszły fazę II badań klinicznych mogą być dostarczane przez producenta bezpłatnie osobom, które nie mają już żadnej alternatywy terapeutycznej wśród dostępnych w tym momencie leków. Dane uzyskane z badań prowadzonych w ramach expanded access mogą stanowić uzupełnienie wyników fazy III.
Po dokładnej analizie danych związanych ze stosowaniem leku u ludzi, gdy stosunek korzyści do ryzyka jest wyraźny, można rozpocząć III fazę badań.
III Faza III faza badań klinicznych trwa od roku do kilku lat, i jest prowadzona z udziałem aż kilku tysięcy chorych (od 1 000 do 3 000 i więcej). Pacjenci kwalifikowani do badań III fazy to chorzy ze wskazaniem do farmakoterapii, kwalifikowani są do badania na podstawie ścisłych kryteriów mających na celu przebadanie leku w populacji zbliżonej do populacji chorych występującej w praktyce medycznej. Prowadzona jest ona zwykle przez wiele ośrodków klinicznych, często w wielu krajach. Liczba pacjentów włączanych do badania uwarunkowana jest wymogami rejestracyjnymi (FDA lub EMEA) oraz statystycznymi (szacowana wielkość próby).
Faza III ma na celu ostateczne potwierdzenie skuteczności badanej substancji w leczeniu danej choroby w większej populacji pacjentów, określenie związku pomiędzy jego bezpieczeństwem, a skutecznością podczas krótkotrwałego i długotrwałego stosowania. Ma ona również potwierdzić wyniki dotyczące skuteczności leku uzyskane w fazie II. Pozwala także na poznanie rzadszych działań ubocznych, zdobycie danych koniecznych do dopuszczenia leku do powszechnego stosowania. Ta część badań może trwać od roku do kilku lat. Zasady prowadzenia III fazy badań (zastosowanie metody podwójnie ślepej próby, losowy dobór pacjentów do poszczególnych grup) są takie same, jak w fazie II, wymaga również częstych wizyt w klinice prowadzącej badania.
W tej fazie badania porównywanie działania badanego leku (LB) z placebo (P), albo z lekiem standardowym (LS), wykonywane może być na trzy sposoby:
- poprzez porównanie dwóch grup chorych, z których jedna otrzymuje LB, a druga P lub LS,
- parami, kiedy jeden chory otrzymuje LB, a drugi w tym samym czasie P lub LS,
- krzyżowo- wtedy, gdy jeden chory najpierw otrzymuje LB, potem P lub LS i znowu LB, a drugi chory leczony jest w odwrotnej kolejności - najpierw P lub LS, potem LB i znowu P lub LS.
Badania tej fazy zbierają dane, które są podstawą do rejestracji produktu leczniczego (faza III a) oraz służą celom marketingowym (faza III b). Po pozytywnym zakończeniu III fazy badań lek może zostać zarejestrowany i wprowadzony do obrotu. Na podstawie wyników fazy: III a oraz III b następuje przygotowanie wniosku o rejestrację nowego produktu leczniczego.
Wszystkie dane uzyskane w czasie badań podstawowych, przedklinicznych oraz klinicznych fazy od I do III są obowiązkowym elementem dokumentacji, wymaganej przez instytucje zajmujące się rejestracją leków. Dokumentacja rejestracyjna może liczyć nawet kilkanaście tysięcy stron. Przed rozpoczęciem procesu badawczo-rozwojowego producent produktu leczniczego konsultuje z wiodącymi, instytucjami rejestracji leków zakres niezbędnych danych, w tym kryteria włączenia i wyłączenia oraz wskazania, aby zminimalizować ryzyko odrzucenia dokumentacji rejestracyjnej z powodu pominięcia istotnych danych.
Faza IVIV faza badań klinicznych dotyczy leków już zarejestrowanych i obecnych w sprzedaży. Ma ona na celu określenie, czy produkt jest bezpieczny we wszystkich wskazaniach zalecanych przez producenta i dla wszystkich grup chorych. IV faza badań dodatkowo weryfikuje wcześniej uzyskane wyniki, w tym ewentualne pojawienie się wcześniej nie zarejestrowanych działań niepożądanych.
W przyszłości opracowywanie nowych leków ma być procesem bardziej zintensyfikowanym. Większy nacisk będzie kładziony na prace nad cząsteczką w fazie przedklinicznej. Według ekspertów, nieuniknione jest poszukiwanie możliwości obniżenia kosztów badań klinicznych, zwłaszcza w fazie III.
Twórca leku najpierw zabezpieczy i uruchomi tzw. „in-life testing”, czyli serię małych, ale ściśle ukierunkowanych badań klinicznych. Na podstawie warunkowego zatwierdzenia leku przez agencję regulacyjną, firmie farmaceutycznej wolno będzie wprowadzić go na rynek udostępniając go wstępnie określonej, wąskiej grupie pacjentów. W rezultacie doprowadzi to do pełnej integracji badań klinicznych z praktyką kliniczną. Badania kliniczne będą częścią normalnej praktyki lekarskiej. W rzeczywistości proces zmiany podejścia do pracy nad nowymi lekami potrwa najbliższe dziesięć, a nawet dwadzieścia lat.
Badania kliniczne leków są niezwykle ważne dla poznawania nowych strategii terapeutycznych, dla zwiększania wiedzy o stosowaniu, skuteczności i działaniach ubocznych leków. Dostępne dane wskazują, że nowy lek wprowadzony na rynek pochłania nakłady ok 3–3, 5 mld złotych, z czego aż ok. 25% przypada na badania przedkliniczne i na pierwsze etapy badań klinicznych, obejmujących poszczególne tzw. fazy od I do IV. Jedynie 1 na 5 testowanych leków pomyślnie przechodzi wszystkie fazy badania. Biorąc pod uwagę ilość preparatów poddawanych testom i długi czas ich przeprowadzania, ta liczba może wydawać się bardzo mała. Należy jednak pamiętać, że każde badanie, nawet zakończone niepowodzeniem wnosi wiele do współczesnej medycyny i być może kiedyś w inny sposób pomoże w walce o zdrowie i lepsze życie.
Autor: Zuzanna KoperwasBibliografia:- Rozporządzenie Ministra Zdrowia z dnia 2 maja 2012 r. w sprawie Dobrej Praktyki Klinicznej (Dz. U. z 2012 r. Nr 489).
- Badania kliniczne. Organizacja. Nadzór. Monitorowanie. Marcin Walter (red.) . Warszawa: Stowarzyszenie na Rzecz Dobrej Praktyki Badań Klinicznych w Polsce, 2004. ISBN 8391913414.
- Marek Czarkowski Zagrożenie, ryzyko i szkoda w badaniach klinicznych Pol. Merk. Lek., 2008, XXV, 146, str. 105-109
- McKenzie R, Fried MW, Sallie R i wsp. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med 1995;339:1093-15.
- Norma ISO 14155 (2003-E)
- Część 1 (14155 – 1): Wymagania zasadnicze (26/02/2003
- Część 2 (14155 – 2): Plan badania klinicznego (15/05/2003)
- Badania kliniczne w Polsce- Główne wyzwania, Listopad 2010
- Projektowanie badań biorównoważności oraz badań klinicznych I, II, III fazy , Teresa Brodniewicz, Marcin Ossowski, Podsumowanie szkolenia, Łódź 23.09.2008
- Medycyna Praktyczna, 1-2 (107-108)/2000, http://www.mp.pl/artykuly/?aid=373http://www.infarma.pl/uploads/media/Zasady_prowadzenia_badan_klinicznych.pdf
- http://www.bcfi-vet.be/nl/nlinfos/nlfolia/05FVN3a.pdf
- http://www.mz.gov.pl/wwwfiles/ma_struktura/docs/projekt_ustawy_obk_24052011.pdf
- http://www.nowinylekarskie.ump.edu.pl/uploads/2011/3/219_3_80_2011.pdf
- http://www.badaniaklinicznewpolsce.pl/
- http://www.gcppl.org.pl/
- http://www.pmrpublications.com/press-releases/337/rynek-badan-klinicznych-w-polsce-rosnie-ale-wolno
- http://bio-etyka.blogspot.com/2010/12/raport-pwc-dotyczacy-badan-klinicznych_12.html
- http://infarma.pl/fileadmin/badania_kliniczne_raport/Badania%20kliniczne%20w%20Polsce%202010.pdf
- http://www.urpl.gov.pl/pozwolenia-na-badania-kliniczne
- http://www.aids.gov.pl/files/wiedza/Badania_kliniczne.pdf
- http://www.astrazeneca.pl/513697/513073?itemId=13416663&nav=yes










Recenzje