W artykule zatytułowanym „Pokaż mi swoje DNA, a powiem Ci czym jesteś” omówiono zagadnienia związane z barkodingiem w kontekście identyfikacji genetycznej organizmów żywych należących do różnych jednostek taksonomicznych, w tym roślin.
Pojęcie barkodingu DNA zakłada, że można szybko i dokładnie określić gatunek w oparciu o amplifikację i sekwencjonowanie krótkich, ujednoliconych regionów DNA genomowego lub mtDNA do klasyfikacji zwierząt. Od niedawna, przez National Institutes of Health (NIH) do identyfikacji genetycznej roślin zaleca się badanie dwóch locus, a mianowicie ganów rbcL oraz matK [1].
Molekularna botanika systematyczna odgrywa coraz istotniejszą rolę w medycynie sądowej. Techniki oparte o badania DNA mają zastosowanie zarówno w sprawach karnych, gdy należy dowieść obecność podejrzanego na miejscu zbrodni, jak i do identyfikacji szczepów marihuany. Do badań porównawczych wykorzystuje się DNA pozyskane z poszczególnych okazów roślinnych znanych gatunków. Bardzo często materiał dowodowy stanowią mieszaniny roślin o nieznanej kompozycji. Identyfikacja genetyczna umożliwia rozdzielenie poszczególnych szczepów roślin oraz dostarcza dodatkowych informacji na podstawie ujawnionych śladów, które do tej pory często były ignorowane. Bazy National Center for Biotechnology Information (NCBI) zawierają liczne sekwencje DNA roślin, które mogą być stosowane do identyfikacji i analizy porównawczej dowodów botanicznych zebranych w trakcie śledztwa. W bazach przechowywane są zarówno sekwencje genów jądrowych ,cpDNA oraz mtDNA [2].
Różne oblicza konopi
Bez wątpienia handel narkotykami stanowi poważny proceder, w którego zwalczanie są zaangażowane urzędy celne oraz inne organy ścigania. Instytucje te przeznaczają ogromne środki na zahamowanie importu i podaży nielegalnych środków odurzających, do których należy również marihuana [3]. Obecne prawo napiętnuje rozpowszechnianie marihuany [3], jednakże nie należy zapominać, że wiele historycznych podań wskazuje na właściwości lecznicze konopi [4].
Cannabis sativa L. (konopie siewne) jest jednym z kilku gatunków roślin o długiej historii uprawy używanej jako medykament. Prawdopodobnie leczenie substancjami zawartymi w konopiach zalecano chorym od co najmniej 10000 lat [5]. Poza mieszkańcami Indii, wartość leczniczą konopi znali również starożytni Chińczycy. Określenie taksonomicznej przynależności gatunków leczniczych występujących w różnych obszarach geograficznych jest problematyczne. Linneusz był zdania, że jest to jeden i ten sam gatunek, natomiast Lamarck ustalił, iż indyjski szczep różni się od gatunków konopi występujących na terenie Europy. W związku z czym dla rozróżnienia, konopiom występującym w Indiach nadał specyficzną nazwę Cannabis indica (konopie indyjskie). Za źródło pochodzenia konopi uważa się Azję Środkową, skąd to następnie rozprzestrzeniły się na obszar krajów śródziemnomorskich, docierając także na teren Europy Wschodniej i krajów Europy Środkowej [6]. Trudno jest określić dokładne źródło pochodzenia roślin ze względu na ich wielowiekową historię upraw. Z pomocą przychodzą badania na poziomie molekularnym. Analiza sekwencji nukleotydowej DNA próbek pobranych w trakcie prowadzenia wykopalisk archeologicznych oraz świeżych próbek roślin dostarcza informacji na temat ewolucji gatunku [4].
Które geny wybrać do analizy?
Najszerzej scharakteryzowanym locus u wszystkich gatunków roślin jest duża podjednostka rybulozo – 1,5 – karboksylazy bisfosforanu (rbcL). Gen rbcL zlokalizowany jest w genomie chloroplastów. Baver i wsp. wytypowali locus rbcL jako układ modelowy do opracowania molekularnej metody identyfikacji gatunkowej roślin. W swoim wyborze kierowali się wysoką liczebnością występowania w komórkach roślinnych oraz obszernością przebadanych sekwencji DNA znajdujących się w bazie danych NIH [2].
Drugim kandydatem, sugerowanym przez Consortium for the Barcode of Life (CBOL) Plant Working Group, do zastosowania w procesie barkodingu jest gen matK (ang. maturase K gene). Gen matK składa się z ok. 1570 par zasad i koduje białko maturazy. Region kodujący matK zazwyczaj znajduje się w obrębie intronu chloroplastowego genu trnK. Badania prowadzone nad analizą genu matK dowodzą, że region ten w wysokim stopniu umożliwia różnicowanie gatunków, Niestety pod względem technicznym, amplifikacja sekwencji kodującej wybrany region DNA jest utrudniona, ze względu na powtarzalność występujących po sobie, pojedynczych nukleotydów [7].
Na całym świecie trwają badania nad licznymi obszarami DNA roślin, na podstawie których, również jest możliwa ich identyfikacja i klasyfikacja. Badania filogenetyczne wielu gatunków oparte są w dużym stopniu na analizie polimorficznych loci DNA chloroplastowego, np. trnL, trnF, intronu tRNA, które to regiony wykazują dużą zmienność w obrębie haplotypów [3], [4].
Przykładowe zastosowanie technik molekularnych
Identyfikacji genetycznej roślin można dokonywać z wykorzystaniem techniki losowej amplifikacji polimorficznego DNA, tzw. RAPD-PCR (ang. Randomly Amplified Polymorphic DNA), która pozwala odróżnić od siebie badane osobniki. Metodę RAPD po raz pierwszy zastosował w 1990 r. Williams i wsp. [9]. Podobnie jak PCR przebiega ona w trzech następujących po sobie etapach, czyli denaturacja matrycy, przyłączanie starterów oraz wydłużanie łańcucha DNA z optymalnie ustalona liczbą powtórzeń. To co odróżnia standardową technikę PCR od RAPD to specyfika doboru oligonukleotydowych starterów. W metodzie RAPD wykorzystuje się pojedynczy, krótki starter o długości od 5 do 15 dowolnie wybranej sekwencji nukleotydów. Taki starter przyłącza się do obu nici DNA w kilku miejscach jednocześnie, co przyczynia się do powstania od kilku nawet do kilkunastu produktów amplifikacji. Podczas gdy, do przeprowadzenia reakcji PCR stosuje się dwa różne startery, gdzie każdy przyłącza się tylko do jednej nici DNA. Wyniki badań uzyskane z wykorzystaniem techniki RAPD-PCR prezentuje się w postaci żeli, po rozdziale elektroforetycznym produktów reakcji losowej amplifikacji polimorficznego DNA. [8]
Przedstawioną powyżej metodę do identyfikacji genetycznej Cannabis sativa wybrali Gillan R. i wps. [10] oraz Jagadish V. i wsp. [11], co można uzasadnić próbą ominięcia problemów związanych ze słabo poznaną sekwencją nukleotydów budujących analizowany region DNA. Jednakże, Linarce i wsp. [3] uznali, że technika ta nie jest wystarczająco miarodajna, ze względu na losowość amplifikowanych fragmentów nici DNA. W badaniach mających na celu wskazanie specyficznych starterów do identyfikacji gatunkowej na podstawie analizy polimorfizmu dwóch chloroplastowych genów: trnL, trnF, zdecydowali się na klasyczną metodę PCR.
Podstawowy materiał do badań nad C. sativa stanowiły liście czterech różnych roślin. Pierwszy okaz pochodził z Indii pozostałe trzy z Afryki Południowej. Wyizolowane DNA z czterech różnych liści stanowiło matrycę reakcji PCR. W pierwszej kolejności DNA amplifikowano ze starterami uniwersalnymi: C oraz F (Tab.1.). Długość uzyskanych produktów wynosiła ok. 818 par zasad, co pokrywa się z dotychczasowymi danymi literaturowymi. Każdy z czterech produktów został zsekwencjonowany. Stopień homologii pomiędzy dwoma okazami C. sativa pochodzącymi z Afryki Południowej wynosił 97,5%. Dodatkowo uzyskane sekwencje nukleotydowe porównano z danymi zgromadzonymi w EMBL Nucleotide Sequence Database (EMBL-Bank). Na podstawie przeprowadzonych analiz wykazano wysoki współczynnik homologi sekwencji C. sativa oraz Nicotinia, wynoszący 68% [3].
Tab.1. Sekwencje starterów użytych do amplifikacji cpDNA [3].
Nazwa startera Sekwencje 5'- 3'
C CGAAATCGGTAGACGCTACG
D GGGATAGAGGGACTTGAAC
E GGTTCAAGTCCCTCTATCCC
F ATTTGAACTGGTGACACGAG
G GAGGGTTTCTAATTTGTTATGTT
H ACTAGAGGACTTGGACTATGTC
Następnie, przeprowadzono łańcuchową reakcję polimerazy z zastosowaniem starterów uniwersalnych oraz specyficznych względem Cannabis sativa: C,D, G i H (Tab.1.). Specyficzne startery opracowano w oparciu o znajomość uprzednio poznane sekwencje genów poddawanych amplifikacji, za pomocą narzędzia bioinformatycznego jakim jest FastA. Wybrano takie regiony DNA C. sativa, których sekwencje odznaczały się niskim stopniem homologii w stosunku do sekwencji innych gatunków roślin. Postępując zgodnie z swoimi założeniami, badacze przeprowadzili reakcje duplex PCR z zastosowaniem zestawu wybranych starterów, używając jako matrycy DNA wyizolowane nie tylko z liści C. sativa, ale także innych gatunków roślin. Wyniki tego etapu badań przedstawiono na Ryc.1. [3].
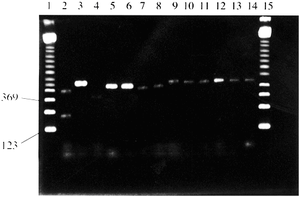
Ryc.1. Wyniki rozdziału elektroforetycznego produktów PCR różnych okazów botanicznych na 2% żelu agarozowym [3].
Wszystkie reakcje PCR były przeprowadzone ze starterami C, D, G i H, poza ścieżką 5, gdzie zastosowano wyłącznie startery C i D. Ścieżki 1 i 15 – marker wielkości 123pz (Sigma); ścieżka 2 - Cannabis sativa; ścieżka 3 - Arabadopsis; ścieżka 4 - Brassica ścieżka 5 - Zea; ścieżka 6 - Zea; ścieżka 7 - Capsicum; ścieżka 8 - Festuca; ścieżka 9 - Ulex; ścieżka 10 - Humulus; ścieżka 11 - Vicia; ścieżka 12 - Paperva; ścieżka 13 - Oryza; ścieżka 14 - Nicotinia.
W doborze pozostałych roślin kierowano się wysokim stopniem pokrewieństwa w oparciu o klasyfikację taksonomiczną. Para starterów CD odpowiada za powielanie fragmentu DNA o długości 348 par zasad, natomiast para starterów GH umożliwia otrzymanie produktu o długości 197 par zasad dla C. sativa. Za pomocą techniki PCR, na bazie DNA pozostałych roślin udało się uzyskać tylko jeden produkt o długości ok. 348 par zasad. Na podstawie przeprowadzonych analiz, autorzy pracy w reakcji łańcuchowej polimerazy upatrują metodę umożliwiającą przeprowadzanie szybkich i pewnych testów identyfikacji genetycznej Cannabis sativa. Należy stosować obie pary starterów, ponieważ brak specyficznego prożka w żelu na wysokości 197 par zasad nie może dać pewności co do wyniku negatywnego. Obecność niespecyficznego produktu o długości ok. 348 par zasad daje podstawy by wnioskować, reakcja przebiegła w sposób prawidłowy. Jego obecność stanowi pewnego rodzaju kontrolę dodatnią [3].
Wyniki prac Linarce i wsp.[3] wykorzystali Mukherjee i wsp. [4] do badań nad próbkami archeologicznymi C. sativa. Najstarsze próbki konopi skrywał jeden z grobów Yanghai w okolicy oazy Turfan (Autonomiczny Region Sinkiang-Ujgur), gdzie złożono zmumifikowane ciało mężczyzny rasy kaukaskiej, w wieku około 40 lat. Nasiona, liście i pędy zostały umieszczone w drewnianej misce i skórzanym koszu obok głowy zmarłego. Wyniki badań przeprowadzonych, z wykorzystaniem mikroskopu elektronowego wskazały że zioła, mimo upływu czasu, zachowały się w doskonałym stanie. Dodatkowo, z obszaru przyległego do wspomnianego grobu, zebrano liście rosnących wokół chwastów zidentyfikowanych jako konopie. Ze wszystkich wymienionych rodzajów materiału wyizolowano całkowite DNA [4].
Warto zauważyć, że w swoich pracach do reakcji PCR z jednej strony, zastosowali startery pierwotnie zaproponowane przez swoich poprzedników (Tab.1.) w celu analizy chloroplastowego DNA, natomiast z drugiej strony wykorzystali sekwencje: ITS4 (50-TCCTCCGCTTATTGATATGC-30) oraz ITS5 (50-GGAAGTAAAAGTCGTAACAAGG-30) pod kątem analizy jądrowego rybosomalnego DNA. Udało się powielić oba rodzaje DNA. Kolejny etap prac polegał na przeprowadzeniu reakcji sekwencjonowania, w celu uzyskania kompletnych informacji na temat uzyskanych. Uzyskano fragmenty DNA o długości 185 oraz 186 par zasad. Sekwencjonowanie zakończone sukcesem świadczy o tym, że materiał genetyczny archeologicznego znaleziska przetrwał próbę czasu w dobrym stanie [4].
BLAST prawdę Ci powie
Na ostatnim etapie analizy, otrzymane sekwencje nukleotydowe, próbek podlegających identyfikacji, porównano z sekwencjami zawartymi w bazie NCBI. W oparciu o przeprowadzone analizy porównawcze, zaobserwowano wysoki stopień podobieństwa pomiędzy uzyskanymi sekwencjami nukleotydowymi konopi, a danymi uprzednio opublikowanymi, głównie w regionie trnL-trnF. Ponadto, wykazano, że materia starożytny oraz świeżo zerwany z rejonu wykopalisk ma to samo podłoże filogenetyczne [4].
Łatwe i szybkie wyszukiwanie analogicznych sekwencji umożliwia program internetowy, jakim jest BLAST. Z pośród tysięcy wcześniej zidentyfikowanych sekwencji roślin, BLAST wskazuje listę tych organizmów, które charakteryzują się najwyższym stopniem dopasowania do sekwencji badanych próbek. Stopień zgodności porównywanych sekwencji może wynosić nawet 100% lub nieznacznie niższy odsetek. Wysoki stopień dopasowania na poziome zbliżonym do wartości 100% sugeruje ewentualną identyfikację na poziomie gatunkowym [2].
Analiza sekwencji DNA roślin stanowi funkcjonalne narzędzie w badaniach filogenetycznych, umożliwiając ustalenie relacji zachodzących pomiędzy różnymi organizmami. Baza danych NCBI zawiera reprezentatywne sekwencje nukleotydowe dla poszczególnych gatunków roślin. Na podstawie wyników uzyskanych w programie BLAST nie zawsze da się jednoznacznie ustalić gatunek, natomiast określenie lub potwierdzenie przynależności do danego rodzaju lub rodziny niejednokrotnie niesie ze sobą dużą wartość [2]. W celu ułatwienia użytkownikom na całym świecie korzystania z możliwości jakie daje program BLAST opracowano instrukcję obsługi aplikacji [12].
Wszelkie działania podejmowane przez badaczy zmierzają do utworzenia szybkiego i pewnego systemu identyfikacji oraz klasyfikacji zarówno gatunków roślinnych jak i zwierzęcych. Analizy materiału biologicznego na poziomie molekularnym stanowią niezastąpione narzędzie w codziennej pracy osób zajmujących się identyfikacją genetyczną. W oparciu o współpracę ze specjalistami różnych dziedzin otwierają się nowe możliwości praktycznego wykorzystania wiedzy.
Autor: Agnieszka Gudek
Literatura:
1. Floyd R., Lima J., deWarad J., Humble L., Hanner R. Common goals: policy implications of DNA barcoding as a protocol for identification of arthropod pests. Biol Invasions, 2010.
2. Bever R., Cimino M. FORENSIC MOLECULAR BOTANY: IDENTIFICATION OF PLANTS FROM TRACE EVIDENCE. International Journal of Legal Medicine, 2009;123: 395-401.
3. Linarce A., Thorpe J. Detection and identification of cannabis by DNA. Forensic Science International, 1998; 91: 71–76
4. Mukherjee A., Roy S. C., De Bera S., Jiang H., Li X., Li H., Bera S. Results of molecular analysis of an archaeological hemp (Cannabis sativa L.) DNA sample from North West China. Genet Resour Crop Evol ,2008; 55: 481–485.
5. Schultes R. E., Klein W. M., Plowman T., Lockwood T. E. Cannabis, an example of taxonomic neglect. Bot Mus Leafl Harv Univ, 1974; 23:337–367.
6. Faeti V, Mandolino G., Ranalli P. Genetic diversity of Cannabis sativa germplasm based on RAPD markers. Plant Breed, 1996; 115: 367–370.
7. Yu J., Xue J.H., Zhou S.L. New universal matK primers for DNA barcoding angiosperms. Journal of Systematics and Evolution, 2011; 49 (3): 176–181.
8. Robak M., Baranowska K., Barszczewski W., Wojtatowicz M. RAPD jako metoda różnicowania i identyfikacji drożdży. Biotechnologia, 2005; 4 (71): 142-155.
9. Williams J. G. K., Kubelic A. R., Livak K. J., Rafalski J. A., Tingey S., Nucl. Acid. Res., 1990; 18, (22): 6531-6535.
10. Gillan R., Cole M.D., Linacre AThorpe J.W., Watson N.D. Comparison of Cannabis sativa by random amplification of polymorphic DNA (RAPD) and HPLC of cannabinoids: a preliminary study, Sci. Justice, 1995; 35: 169–177.
11. Jagadish V., Robertson J., Gibbs A. RAPD analysis distinguishes Cannabis sativa samples from different sources, Forensic. Sci. Int. 1996; 79: 113–121.
12. Camacho C., Madden T., Coulouris G., Ma N., Tao T., Agarwala R., Morgulis A. BLAST Command Line Applications User Manual. Link:
http://www.ncbi.nlm.nih.gov/books/NBK1763/?campaign=facebook-09132012
https://laboratoria.net/home/15651.html