
|

Zamknij X
|





 Zrealizowanie projektu dotyczącego poznania ludzkiego genomu umożliwiło naukowcom zsekwencjonowanie wszystkich genów występujących w DNA. Zamierzeniem tych badań było zrozumienie funkcjonowania organizmu ludzkiego. Poznanie genomu umożliwiło zdobycie wiedzy o białkach kodowanych przez DNA, jednak nadal nie zostały wyjaśnione mechanizmy działania komórek, organów oraz całego organizmu. Ocenia się, że w genomie ludzkim jest zapisanych około 35 tysięcy genów kodujących 200-300 tysięcy białek. Poznano strukturę oraz funkcje nieznacznej części tych białek.
Zrealizowanie projektu dotyczącego poznania ludzkiego genomu umożliwiło naukowcom zsekwencjonowanie wszystkich genów występujących w DNA. Zamierzeniem tych badań było zrozumienie funkcjonowania organizmu ludzkiego. Poznanie genomu umożliwiło zdobycie wiedzy o białkach kodowanych przez DNA, jednak nadal nie zostały wyjaśnione mechanizmy działania komórek, organów oraz całego organizmu. Ocenia się, że w genomie ludzkim jest zapisanych około 35 tysięcy genów kodujących 200-300 tysięcy białek. Poznano strukturę oraz funkcje nieznacznej części tych białek.
Prowadzone są liczne badania dotyczące właściwości wszystkich białek występujących w organizmie człowieka. Taki kompletny zestaw białek danego organizmu nazwano proteomem (z ang. protein complement of the genome), co oznacza komponent białkowy kodowany przez genom. Dyscypliną zajmującą się proteomem jest szybko rozwijająca się dziedzina nauki zwana proteomiką. Pojęcie proteomiki, czyli analizy proteomu wprowadzono w połowie lat 90.XX wieku. Celem proteomiki jest identyfikacja, a także poznanie struktury, funkcji oraz oddziaływań jakie są zakodowane w genomie białek tworzących proteom. Istotny jest fakt, że wszystkie komórki danego organizmu mają wspólny genom, jednak w poszczególnych typach komórek i tkanek jego różne frakcje ulegają ekspresji. Badania proteomiczne skupiają się na dwóch obszarach badań. Pierwszy jest związany z ekspresją białek, natomiast drugi z ich wzajemnymi oddziaływaniami. Celem proteomiki ekspresji jest poznanie wzorów ekspresji białka w określonych warunkach, jak również dążenie do ustalenia odpowiedniej mapy całego proteomu. Użycie takiej mapy miałoby znaczenie w badaniach toksykologicznych, projektowaniu nowych leków, a także przy identyfikacji biomarkerów. Biomarkerami są białka, których poziom ekspresji umożliwia dostarczenie informacji o wystąpieniu określonych chorób, bądź skuteczności działania leku. Analiza profili białkowych osób zdrowych i chorych, których celem jest wykrycie różnic pomiędzy nimi należy do podstawowych zadań proteomiki klinicznej. Naukowcy w przeprowadzanych badaniach przeszukiwania całego proteomu osoby chorej zauważają odchylenia. Należy do nich odmienna ekspresja białek aniżeli w warunkach fizjologicznych. Białka te zostają biomarkerami określonego stanu patologicznego. O wyborze metody zastosowanej w proteomice decyduje wiele parametrów białek, na przykład poziom ich ekspresji i modyfikacji, lokalizacji wewnątrz komórek, a także złożoności ich mieszanin. Podział technologii proteomicznych można podzielić na techniki stosowane do charakterystyki białek i generowania map białkowych jak również takie, które służą do badań funkcji białek oraz interakcji między nimi. Pierwszą grupę proteomicznych narzędzi stanowi elektroforeza dwuwymiarowa, spektrometria mas oraz techniki, które pozwalają na identyfikację białek takie jak „białkowy odcisk palca”. W badaniach interakcji przede wszystkim stosuje się metody oparte na immunopowinowactwie, a także drożdżowy system dwuhybrydowy. Na współczesną proteomikę składają się badania podstawowe oraz kliniczne. Pomiędzy tymi dyscyplinami występuje ciągła wymiana informacji, tak zwana translacja co zasadniczo wpływa na trwały postęp wiedzy o danych schorzeniach [1].
Znaczenie spektrometrii mas w proteomice klinicznej
W badaniach proteomicznych łączy się metody biochemiczne, fizykochemiczne i bioinformatyczne. W badaniach tym możliwe jest jednoczesne analizowanie tysięcy białek z danego typu komórek lub tkanek. Do lat 70-tych XX w. masy białek określano za pomocą takich metod jak elektroforeza, ultrawirowanie czy chromatografia. Metody te były jednak niedokładne, błąd wynosił 10-100%. Dopiero wprowadzenie w latach 90-tych techniki spektrometrii mas (ang. Mass Spectrometry MS) przyczyniło się do szybkiego postępu w tej dziedzinie, dzięki wdrożeniu nowych technik jonizacji. MS jest zdecydowanie konkurencyjna w stosunku do innych metod na ogół stosowanych w proteomice, przede wszystkim z uwagi na możliwość analizy białek będących w bardzo małych stężeniach, a także w skomplikowanych mieszaninach. Spektrometria masowa jest techniką analityczną pozwalającą na precyzyjny pomiar stosunku masy do ładunku elektrycznego jonu, gdzie przy znanym ładunku jonu daje możliwość obliczenia masy z dokładnością do pojedynczych atomów. Typowy spektrometr masowy składa się z komory jonizacyjnej, analizatora różnicującego jony pod względem stosunku ich masy do ładunku i detektora zliczającego liczbę jonów, które są sprzężone z komputerowym systemem rejestracji i analizy danych (Rys. 1) [2].
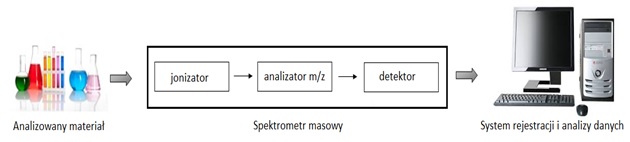
Spektrometria masowa znalazła zastosowanie w badaniach budowy strukturalnej cząsteczek, składu chemicznego, czystości danej substancji oraz identyfikacji zanieczyszczeń. Głównymi zaletami spektrometrii masowej są dokładność wyznaczenia masy, rozdzielczość do kilku jednostek masy atomowej oraz szeroki zakres stosowania, od peptydów do dużych białek (>200 kDa). Ponadto MS umożliwia detekcję strukturalnych wariantów białek (białka zmodyfikowane posttranslacyjnie, mutanty). Znaczenie spektrometrii mas polega na rozdzielaniu w polu elektromagnetycznym ze względu na wartość stosunku masy m do ładunku z (m/z) zjonizowanych cząstek, jakie znajdują się w analizatorze w wysokiej próżni. Zależnie od rodzaju analizowanych substancji, używa się różnych technik jonizacji i rozdziału jonów. W MS do analiz białek wykorzystywane są niskorozdzielcze analizatory (kwadrupolowe, pułapki jonowe), bądź wysokorozdzielcze (mierzące czas przelotu jonów, cyklotronowy rezonans jądrowy z transformacją Furiera). Najbardziej rozpowszechnionymi metodami jonizacji są desorpcja laserem w stałej matrycy MALDI (ang. matrix-assisted laser-desorption ionization) oraz elektrorozpylanie (ESI, ang. Electrospray) [3].
Technika ESI polega na rozpylaniu cieczy, która zawiera badaną substancję, w polu elektrycznym o wysokim napięciu (około 1-5 kV). Na ogół metoda ta nie powoduje fragmentacji badanych cząsteczek. Podczas wykorzystania tej techniki wzbudzania, powstające jony często mają ładunek wielokrotny, natomiast liczba przyłączonych protonów zależna jest od liczby zasadowych aminokwasów jakie występują w badanych cząsteczkach. Cząsteczka białka ma możliwość przyłączenia kilkunastu, a nawet kilkudziesięciu protonów dlatego można analizować białka o wysokich masach cząsteczkowych, które mogą wynosić kilkaset tysięcy daltonów (Da). Głównymi zaletami tej metody są: wysoka czułość oznaczeń, możliwość analizy dużych cząsteczek do około 80 000 Da, minimalna fragmentacja próbki w czasie jonizacji, a także kompatybilność z technikami chromatograficznymi i elektroforetycznymi. Dzięki sprzężeniu ESI, a w późniejszym czasie także MALDI, z chromatografią cieczową uzyskano dalsze zwiększenie czułości analiz w MS [3].
Inną metodą jonizacji stosowaną w spektrometrach służących do analizy peptydów i białek jest desorbcja laserowa w stałej matrycy - MALDI.W technice tej stosuje się jonizację wiązką laserową o odpowiednio dobranej energii, by nie doprowadzać do fragmentacji cząsteczek, a tylko do ich „wybijania” z właściwie przygotowanej matrycy. Matryca pochłania energię lasera, która z kolei przekazywana jest do analizowanych cząsteczek. Spektrometry MALDI stosowane są do analizy białek o masach od ~1000 Da do nawet kilkuset tysięcy Da. Technika MALDI polega na kokrystalizacji cząstek substancji badanej z cząsteczkami matrycy absorbującej światło, zwykle kwasów aromatycznych. Zazwyczaj jako matryce wykorzystuje się pochodne kwasu cynamonowego (np. kwas synapinowy, kwas α-cyjano-4-hydroksycynamonowy). Cząsteczki białka w matrycy wzbudza się za pomocą światła lasera. W wyniku tego z kryształów desorbowane są uprotonowane cząsteczki próbki, a powstałe kationy, mające na ogół pojedynczy ładunek ([M + H]+), które nazywane są jonami molekularnymi. W normalnych warunkach nie obserwuje się ich fragmentacji.W podobny sposób działają spektrometry typu SELDI (ang. surface-enhanced laser desorption ionization). Wykorzystują one desorbcję zjonizowanych światłem laserowym cząsteczek białek następujących z odmiennego typu powierzchni swoiście wiążących białka. Takie powierzchnie są pokryte substancjami będącymi odpowiednikami złóż chromatograficznych. SELDI stosowane są przede wszystkim w proteomice klinicznej w badaniach przesiewowych. Powszechne zastosowanie proteomiczne mają typy analizatorów m/z - czasu przelotu jonów – ToF (ang. time of flight), które rejestrują czas przelotu zjonizowanych cząsteczek (zazwyczaj 0,01-1 ms) i są odwrotnie proporcjonalne do wartości m/z analizowanych jonów. Z widma masowego można odczytać jaka liczba jonów o precyzyjnie określonej masie została zliczona przez detektor [3].
Spektrometria mas wykorzystywana jest także do badań strukturalnych białek, a szczególnie do ich identyfikacji. W tym celu oczyszczone białko początkowo poddaje się trawieniu na mniejsze fragmenty przy pomocy enzymu (zastosowanie znajdują tu proteazy, jak: chymotrypsyna, trypsyna oraz kolagenoza). Uzyskane peptydy wymywa się z żelu, a następnie poddaje analizie. Na spektrometrze MALDI-TOF otrzymuje się widmo, które ukazuje masy poszczególnych poptydów. Jest to mapa peptydowa, typowa dla każdego białka, co jest skutkiem specyfiki substratowej enzymu. Jako przykład można podać trypsynę, która zrywa wiązania w białku zawsze po resztach aminokwasowych lizyny i argininy, natomiast masy uzyskanych fragmentów zależne są od pozycji tych dwóch aminokwasów w sekwencji białkowej. W wyniku trawienia otrzymane masy peptydów są porównywane z masami peptydów wynikającymi z trawienia in silico sekwencji aminokwasowych, które są obecne w białkowych bazach danych. Metoda ta umożliwia jedynie identyfikację poznanych wcześniej białek [2].
Inna metoda identyfikacji białek i peptydów, tandemowa spektrometria mas z kolizyjnie indukowaną fragmentacją - CID MS/MS, umożliwia ich jednoczesne sekwencjonowanie, za pomocą zastosowania spektrometrów z podwójnym analizatorem. W pierwszym analizatorze selekcjonuje się wybrany jon molekularny bądź fragmentacyjny spośród powstających w czasie procesu wzbudzania jonów. Jon fragmentacyjny poddawany jest w komorze kolizyjnej zderzeniom z atomami wprowadzonego gazu szlachetnego (hel albo argon), co skutkuje rozpadem jonu macierzystego na fragmenty (jony potomne). W oparciu o masę jonów potomnych jest ustalany skład aminokwasowy danego peptydu, którego jony poddane zostały zderzeniom w komorze kolizyjnej. Prawidłowa analiza kombinatoryczna widma jonów potomnych umożliwia ustalenie sekwencji aminokwasów w łańcuchu peptydowym. Wykorzystanie takiego podejścia daje możliwość identyfikacji nieznanych wcześniej białek. Wysokorozdzielcze spektrometry mas na przykład z analizatorami wykorzystującymi zjawisko cyklotronowego rezonansu jądrowego z transformacją Fouriera - FT-ICR pozwalają na wyznaczenie masy cząsteczkowej z dokładnością 10-4. Daje to możliwość określenia podstawowego składu analizowanego jonu z dużą precyzją [2].
Aktualnie popularnością cieszą się podejścia metodyczne, w których wykorzystuje się sprzężenie aparatów do chromatografii cieczowej ze spektrometrem mas (LC-MS). W tym przypadku rozdziela się metodami chromatografii cieczowej mieszaniny białek albo peptydów uzyskanych po enzymatycznym trawieniu białek, a kolejnie analizuje się w różnego typu spektrometrach mas. Eluat z kolumny chromatograficznej jest bezpośrednio wprowadzany do komory jonizacyjnej spektrometrów ESI, bądź też frakcje chromatograficzne są zbierane i kokrystalizowane z matrycą na prawidłowych płytkach, a następnie analizowane w spektrometrach MALDI-TOF. Dwuwymiarowa analiza, gdzie pierwszy wymiar stanowi czas retencji na kolumnie chromatograficznej, natomiast drugim wymiarem jest wartość m/z przyczynia się do jednoczesnej detekcji w mieszaninie kilkuset białek [2].
Podstawowe zadania proteomiki klinicznej
Podstawowym zadaniem proteomiki klinicznej (ang. clinical proteomics) jest odkrywanie różnic w proteomie osób zdrowych oraz chorych, związków pomiędzy stanem proteomu a stanem rozwoju choroby, a także zmian zachodzących w profilach białkowych inicjowanych w odpowiedzi na czynnik terapeutyczny. Takie czynniki nazywane są biomarkerami bądź znacznikami, a ich stan oraz ilość wskazuje na ryzyko choroby, procesy zawansowania choroby, a także efekty odpowiedzi na interwencję terapeutyczną. Tak więc celem proteomiki klinicznej jest identyfikacja oraz scharakteryzowanie biomarkerów białkowych, które są wykorzystywane w molekularnej diagnostyce choroby [4].
W diagnostyce medycznej przedmiotem badań proteomiki klinicznej, może być tkanka bądź organ docelowy, w którym następuje rozwój choroby, lub tkanka zastępcza. Najczęściej jest to krew albo materiał pochodny (osocze bądź surowica). We krwi znajdują się komórki oraz produkty ich metabolizmu, zarówno te w których rozwija się proces chorobowy jak i takie które mają styczność z chorym narządem bądź tkanką (np. komórki układu odpornościowego). Dokonując analizy surowicy lub osocza krwi można otrzymać wzór zestawu białek i peptydów, charakterystyczny dla danego osobnika (profil białkowy, widmo masowe). Następnie, stosując metody analizy matematycznej, porównywane są profile charakterystyczne dla osób zdrowych i chorych. Jednocześnie poszukiwane są różnicujące je „piki”. W najlepszym układzie należy zidentyfikować obecność białek w takich różnicujących „pikach”, natomiast zmiany poziomu zidentyfikowanego biomarkera sprawdza się innymi metodami (np. immunologicznymi) [5].
Jednym z możliwych zastosowań analizy proteomu krwi przy użyciu spektrometrii mas jest molekularna diagnostyka nowotworów. Przyczyną, jak również konsekwencją choroby nowotworowej, mogą być zmiany profilu peptydów i białek krwi. Bez względu na to powinny one przedstawiać patologiczny stan narządu oraz całego organizmu, co umożliwa diagnostykę choroby. Obecnie poszukuje się zestawu znaczników, w których każdy składnik osobno posiada niewielką przydatność diagnostyczną, lecz jako kompletny zestaw mają wysoką swoistość i czułość. Panel markerów, mający kilka bądź kilkadziesiąt składowych, nazywany jest klasyfikatorem. Posiadają one poziomy ekspresji wybranych genów wyselekcjonowane za pomocą metod funkcjonalnej genomiki. Zakłada się, że analiza wieloskładnikowych zestawów białek oraz peptydów wybranych ze złożonych profili proteomicznych surowicy krwi będzie posiadać znaczenie w praktykach klinicznych [6]. Analiza proteomu tkanki docelowej jest o wiele wiarygodniejszym źródłem informacji o chorobie nowotworowej aniżeli badanie tkanki zastępczej. Jednakże w badaniach przesiewowych, wczesnego wykrywania choroby w stadium „przedklinicznym", wykrywania mikroprzerzutów bądź diagnostyki nowotworów leczonych bez zastosowania chirurgii, analiza proteomu krwi jest niezbędną metodą diagnostyczną [7].
Zastosowanie analizy proteomicznej krwi w diagnostyce chorób nowotworowych
Analizę proteomu surowicy krwi osób z chorobą nowotworową można przeprowadzić za pomocą metod spektrometrii mas. Pierwszą pracę w tej dziedzinie opublikował zespół Petricoin i Liotta w 2002 roku. Zastosowali oni metodę SELDI-TOF do identyfikacji w surowicy krwi peptydów swoistych dla chorych na nowotwór jajnika. Ostatnie lata obfitują w publikacje dotyczące przydatności technik spektrometrii mas, głównie MALDI-ToF i SELDI-TOF, do analizy niskocząsteczkowej frakcji peptydów surowicy (sporadycznie osocza) pacjentów z chorobą nowotworową. Badania profili białkowych surowicy krwi dały możliwość identyfikacji klasyfikatorów (wieloskładnikowych markerów białkowych), które mają zastosowanie we wczesnym wykrywaniu nowotworów i ich diagnostyki. Spektrometria mas znalazła zastosowanie do analizy proteomu surowicy krwi w diagnostyce nowotworów regionu głowy i szyi, piersi, jajnika, macicy, prostaty, płuc, jelita grubego, trzustki, tarczycy, nerki, pęcherza oraz wątroby. Badania zmian profilów masowych białek surowicy u chorych objętych terapią przeciwnowotworową mogłyby wpłynąć na identyfikację markerów molekularnych, które umożliwiają monitorowanie odpowiedzi pacjenta na zastosowaną terapię (np. jej skuteczność, toksyczność) [7].
Analiza profilu białkowego wykonanego za pomocą spektrometru MALDI-ToF (Rys.2) ma wiele zalet jako metoda analityczno-diagnostyczna. Posiada możliwość bezpośredniej analizy złożonych mieszanin oraz analizy wielu prób w krótkim czasie i w standardowych warunkach. Można założyć, że w niedalekiej przyszłości metoda ta stanie się jedną z podstawowych metod molekularnej diagnostyki nowotworów [7].
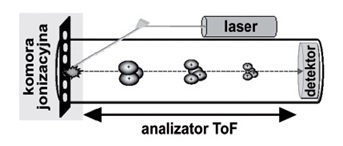
Rys. 2. Schemat budowy spektrometru MALDI-ToF [15].
W wielu pracach poświęconych analizie proteomu krwi osób z chorobą nowotworową zastosowana została analiza widm masowych poprzedzona rozdziałem chromatograficznym mieszaniny tzw. metoda LC-MS [8]. Pozwala ona na zwiększenie czułości analizy i scharakteryzowanie białek obecnych we krwi w stężeniach poniżej 10-12 M. Natomiast sprzężenie chromatografu cieczowego z tandemowym spektrometrem mas LC/MS/MS daje możliwość bezpośredniej identyfikacji rozdzielanych białek, a co za tym idzie tworzenia szczegółowych map białek surowicy. Na podstawie analizy aktualnych danych wynika, że obfitym źródłem potencjalnych markerów jest frakcja peptydów i białek mających niskie masy molekularne, mniej niż kilkanaście tysięcy Da. Białka i peptydy o masach poniżej 20-30 kDa usuwane są z krwioobiegu poprzez filtrację w nerkach, toteż są nietrwałymi znacznikami stanu fizjologicznego organizmu. Oczywiste jest, że te niewielkie białka i peptydy są często związane w kompleksach z innymi białkami surowicy, głównie albuminą, co wpływa na zwiększenie czasu ich obecności w krwiobiegu. Taką analizę wykonano w przypadku krwi pacjentek z rakiem jajnika. Pozwoliła ona na rozpoznanie w frakcji surowicy swoistych fragmentów białek aktywnych w procesie nowotworzenia, na przykład fragmentu białka BRCA2 [9].
Rozwój metod spektrometrii mas umożliwia identyfikację różnorodnych białek. Wykazano, że większość zidentyfikowanych biomarkerów procesu chorobowego to białka, bądź ich fragmenty, które fizjologicznie występują w surowicy w wysokich stężeniach, na przykład fibrynogen i fibrynopeptydy, hemoglobiny, amyloid A czy apolipoproteina A [10]. Odkrycie, że pośród potencjalnych biomarkerów nowotworzenia obecne są swoiste fragmenty „fizjologicznych” białek surowicy, skierowało uwagę badaczy na potrzebę zbadania niskocząsteczkowej frakcji peptydów surowicy poniżej 3000 Da. Przeprowadzono analizę oraz identyfikację 61 peptydów surowicy różnych pacjentów z trzema rodzajami nowotworów (raki piersi, prostaty, pęcherza) oraz ludzi zdrowych. Klasyfikator składający się z 61 peptydów odróżnił pacjentów zdrowych od chorych, a także zróżnicował osoby chore z różnymi rodzajami nowotworów. Świadczy to o tym, że klasyfikator był znacznikiem swoistym rodzaju nowotworu [11].
Zagadnieniem związanym z charakterystyką biomarkerów decydującym o ich przydatności w praktyce klinicznej jest również czułość i swoistość. Czułość markera zdefiniowana jest jako stosunek, wyrażany w procentach, prawidłowo zidentyfikowanych za jego pomocą próbek od osób chorych. Natomiast swoistość określa liczbę próbek od osób zdrowych błędnie zidentyfikowanych za jego pomocą jako chorzy. W przypadku kilkuskładnikowych markerów białkowych, wybranych poprzez analizę widm masowych białek surowicy, czułość, a także swoistość były bardzo wysokie, wielokrotnie przekraczały one 90%. Istotny jest fakt, że wybrane markery peptydowe często nie pozwalają na rozróżnienienowotworów w różnych stopniach zaawansowania. Pomimo tychwątpliwości trudno zlekceważyć korzyści diagnostycznepłynącez wykorzystania markerów peptydowych. Wykorzystanie panelupeptydów zidentyfikowanego metodami spektrometrii mas w kombinacjiz tradycyjnymi markerami zasadniczo podnosi wartośćdiagnostyczną badania. Jako przykład można podać połączoną analizę kilku białek wybranychna podstawie analizy MS oraz „tradycyjnego” markeraCA125 (cancer antigen 125 kDa). Kombinacja taka poprawia wykrywalność wczesnych stadiów rakajajnika [12].
Badania proteomiczne w dużej mierze dotyczą także analizy komórek i tkanek nowotworowych. W tym przypadku zastosowanie spektrometrii mas polega na identyfikacji białek z komórek nowotworowych po ich rozdziale metodami elektroforetycznymi. Podejmowane są także próby użycia spektrometrii mas jako metody obrazowania molekularnego tkanki nowotworowej. W tej metodzie (zwanej imaging MS) w spektrometrach TOF jonizowane są białka, które znajdują się na powierzchni komórek bądź skrawków histologicznych. Następnie sporządza się widma masowe białek preparatu w różnych pozycjach. Natomiast profile białkowe korelowane są z obrazem mikroskopowym. Taką analizę można stosować do klasyfikacji nowotworów, co ma istotne znaczenie w diagnostyce klinicznej [13].
Znaną techniką jest również mikrodyssekcja laserowa, która służy do izolowania z otoczenia poszczególnych komórek. Izolowane tą metodą komórki nowotworowe mogą być analizowane także technikami spektrometrii mas. W wyniku wysokiej czułości spektrometrów analizę typu LC-MS można realizować na materiale pochodzącym z kilku tysięcy „wyciętych” komórek (odpowiada to kilkuset nanogramom całkowitego białka). W prostszej metodzie SELDI-TOF powtarzalne profile białkowe uzyskuje się nawet dla kilkudziesięciu-kilkuset „wyciętych” komórek [14].
Wprowadzenie technologii proteomicznych i ich ciągłe udoskonalanie daje nadzieję na ich zastosowanie w diagnostyce chorób. Intensywny rozwój technik badawczych i ich zastosowanie w połączeniu z uniwersalną metodą spektrometrii mas, w najbliższych latach, daje szansę na dostarczenie potencjalnych metod analitycznych. Czy trwale zaistnieją w metodach proteomiki klinicznej i molekularnej diagnostyki nowotworów, zależeć będzie od możliwości ich wprowadzenia do praktyki medycznej [7].
Podsumowanie
Wiąże się duże nadzieje z rozwojem proteomiki klinicznej, której zadaniem jest analiza zmian proteomu odzwierciedlającego rozwój procesu chorobowego, jak również efektów terapii. Tak więc proteomika kliniczna uzupełnia diagnostykę pacjenta i wspomaga jego leczenie dzięki doborowi najlepszych metod terapeutycznych. Nie wyklucza się możliwości zastosowania w przyszłości zintegrowanych mikrosystemów analitycznych, które umożliwią szybką analizę profilu białkowego oraz uzupełnią identyfikację wad genetycznych. Bez wątpienia metody stosowane w proteomice klinicznej wymagają jeszcze wielu usprawnień jak i standaryzacji. Wymienić tu należy sposoby doboru, przygotowania oraz pobierania prób, metody wstępnego rozdziału białek, a także ilościowego i jakościowego oznaczenia ich składu oraz dobór prawidłowych sposobów przeszukiwania baz danych, analiz bioinformatycznych i statystycznych. Do praktyki klinicznej nowatorskie metody diagnostyczne mogą być wprowadzone wtedy, gdy ich użyteczność będzie wykazana w obszernych, dobrze zaprojektowanych i kontrolowanych badaniach weryfikujących. Wymaga to jednak wysiłku, cierpliwości i dokładnych sposobów normalizacji.
Nowym, dobrze zapowiadającym się działem proteomiki klinicznej jest badanie proteomu surowicy (lub osocza) krwi metodami spektrometrii mas. Istotną cechą nowoczesnych spektrometów mas jest ich duża dokładność i czułość, dająca możliwość analizy cząsteczek obecnych w złożonych mieszaninach w stężeniu poniżej 10~12 M. Umożliwia to poszukiwanie potencjalnych markerów nowotworzenia w tak skomplikowanej mieszaninie, jaką jest surowica krwi. Metoda ta może być także stosowana samodzielnie i być używana w wspomaganiu wykrywania i diagnostyki nowotworów oraz monitorowaniu ich terapii.
Techniki spektrometrii mas znajdujące zastosowanie w proteomice klinicznej mają także ograniczenia. Metoda spektrometrii mas nie jest metodą ilościową, wzrost „piku” w widmie masowym nie musi być bezpośrednio adekwatny wzrostowi stężenia cząsteczek. Ważnym problemem, w związku z wieloetapową procedurą przygotowania próbek do analizy w spektrometrach MALDI i SELDl, jest standaryzacja metodyki między laboratoriami, przede wszystkim wtedy gdy wyniki mogą mieć nie tylko jakościowy charakter. Przez spektrometr mas są rejestrowane wszystkie rodzaje cząsteczek znajdujące się w płynach ustrojowych. W surowicy krwi podstawowymi wykrywanymi białkami są albumina i immunoglobuliny. Obecność albuminy (stanowiącej ok. 60% wszystkich białek surowicy) jest czynnikiem mającym wpływ na jakość analizy białek i peptydów będących celem badania. Usunięcie jej z preparatu przed wykonaniem analizy stanowi poważny problem metodyczny. Kłopotem metody jest też ogromna liczba danych generowanych w jednej analizie, przez co istnieje konieczność stosowania zaawansowanych technik analitycznych oraz matematycznych. Pomimo tych ograniczeń spektrometr masowy posiada wiele zalet. Pozwala na bezpośrednią analizę złożonej mieszaniny białek, posiada wysoką czułością i sporą tolerancją na zanieczyszczenia w analizowanych próbkach. Ponadto wiele prób może być analizowanych w niedługim czasie w standardowych warunkach a koszt pojedynczej analizy nie jest wysoki. Istnieje hipoteza, że metody proteomiki klinicznej na stałe będą zasadniczym zestawem metod diagnostyki molekularnej i ułatwią lekarzom podejmowanie należytych decyzji diagnostycznych i terapeutycznych.
Autor: Katarzyna Czuba
Literatura:
1.Tyers M., Mann M. From genomics to proteomics. Nature. 2003. 422, 193-197.
2.Aebersold R., Mann M. Mass spectrometry - based proteomics. Nature. 2003. 422, 198-207.
3.Mazurkiewicz R. Spektrometria masowa, w: Metody spektroskopowe i ich zastosowanie do identyfikacji związków organicznych. WNT. 2000. 436-537.
4.Azad N.S., Rasool N., Annunziata C.M., Minasian L., Whiteley G., Kohn E.C. Proteomics in clinical trials and practice. Mol. Cell Proteomics. 2006. 5, 1819-1829.
5.Ahmed N., Oliva K.T., Barker G., Hoffmann P., Reeve S., Smith I.A., Quinn M.A., Rice G.E. Proteomic tracking of serum protein isoforms as screening biomarkers of ovarian cancer. Proteomics. 2005. 5, 4625-4636.
6.Liotta L.A., Ferrari M., Petricoin E.F. Clinical proteomics: written in blood. Nature. 2003. 425, 905.
7.Petricoin E.F., Ardekani A.M., Hitt B.A., Levine P.J., Fusaro V.A., Steinberg S.M., Mills G.B., Simone C., Fishman D.A., Kohn E.C., Liotta L.A. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002. 359, 572-577.
8.Martosella J., Zolotarjova N., Liu H., Nicol G., Boyes B.E. Reversed-phase high-performance liquid chromatographic prefractionation of immunodepleted human serum proteins to enhance mass spectrometry identification of lower-abundant proteins. J. Proteome Res. 2005. 4, 1522—1537.
9.Lowenthal M.S., Mehta A.I., Frogale K., Bandle R.W., Araujo R.P., Hood B.L., Veenstra T.D., Conrads T.P., Goldsmith P., Fishman D., Petricoin E.F., Liotta L.A. Analysis of albumin-associated peptides and proteins from ovarian cancer patients. Clin. Chem. 2005. 51, 1933-1945.
10. Hortin G.L. The MALDI mass spectrometric view of the plasma proteome and peptidome. Clin. Chem. 2006. 52, 1223-1237.
11.Villanueva J., Shaffer D.R., Philip J., Chaparro C.A., Erdjument-Bromage H., Olshen A.B., Fleisher M., Lilja H., Brogi E., Boyd J., Sanchez-Carbayo M., Holland E.C., Cordon-Cardo C., Scher H.I., Tempst P. Diffrential exoprotease actvities confer tumor-specific serum peptidome patterns. J. Clin. Invest. 2006. 116, 271-284.
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje