Wybrane metody izolacji kwasu rybonukleinowego (RNA)Intensywne badania kwasu rybonukleinowego (RNA) rozpoczęto jeszcze przed „erą DNA”. Pierwszym etapem badań RNA była jego izolacja i oczyszczanie [4]. Aktualnie istnieje wiele różnych metod izolacji RNA, różnią się one wydajnością i jakością, a także stosowanymi odczynnikami i czasem trwania izolacji. Pomimo szerokiego wyboru izolacji, metody te ciągle są ulepszane i modyfikowane. Głównym zadaniem przy opracowywaniu nowych procedur stało się ich uproszczenie, przy jednoczesnym maksymalnym zabezpieczeniu wyizolowanego RNA przed degradacją [4].
Po raz pierwszy udało się wyizolować RNA z tkanek zatopionych w parafinie w 1991 roku. Pomimo, iż istnieje wiele różnych modyfikacji metod izolacji, nadal możliwe jest uzyskanie jedynie krótkich fragmentów RNA, Jednakże dzięki rozwojowi technik amplifikacji, udało się przybliżyć czułość reakcji do tych, które wykorzystują jako matryce RNA izolowane z tkanek świeżych lub mrożonych [2].
Wcześniejsze badania Ruppa’a i Locker’a wykazały, że RNA ekstrahowane z utrwalonych w formalinie, a następnie zatopionych w bloczkach parafinowych, jest odpowiednim materiałem do badania determinacji ekspresji genów. RNA izolowane z tkanek FPPE (formalin-fixed, paraffin-embedded) może być złej jakości, gdyż jego rozległa degradacja może wystąpić jeszcze przed zakończeniem procesów utrwalania. Ponadto, utrwalanie w formalinie powoduje powstawanie wiązań krzyżowych między kwasami nukleinowymi a białkami [8].
 Słowa kluczowe: izolacja RNA, formalina, bloczki parafinowe, drożdże Saccharomyces cerevisiae, RNAzy ,DNAzy, metoda FFPE, metoda AGPC.
Słowa kluczowe: izolacja RNA, formalina, bloczki parafinowe, drożdże Saccharomyces cerevisiae, RNAzy ,DNAzy, metoda FFPE, metoda AGPC. W komórkach prokariotycznych i eukariotycznych wyróżnia się 3 główne rodzaje RNA. Są to mRNA (informacyjny RNA), tRNA(transportowy RNA) oraz rRNA(Rybosomalny RNA) [4], [9], [10].
Rybosomalny RNA występuje w komórce w największej ilości. Stanowi on 75% komórkowego RNA. rRNA składa się z 3 fakcji, które różnią się stałą sedymentacji. I tak jest to : 25-28S, 18S, 5S i 5,8S. Frakcje te występują w rybosomach eukariotycznych. Z kolei w rybosomach prokariotycznych wyróżnia się: 23S, 16S oraz 5S. Cząsteczki transportowego RNA tj. tRNA, odpowiedzialne są za dostarczanie odpowiednich aminokwasów do rybosomów w czasie syntezy białek. Cząsteczki tRNA są małe, zwykle rzędu do 25kDa. Informacyjne RNA(mRNA) koduje z kolei białka. Ten rodzaj RNA stanowi ok. 1-5% całkowitego RNA w komórce [4], [9], [10].
Wśród metod izolacji RNA z różnych materiałów wyjściowych, wyróżnia się:
1) Izolację specyficznego RNA (poprzez frakcjonowanie całkowitego RNA komórki)
2) Bezpośrednią izolację specyficznego RNA (ograniczona jest ona wyłącznie do tych komórek, które syntetyzują określony RNA w zwiększonej ilości)
3) Izolacja Poli(A)RNA
4) Izolacja RNA z polirybosomów
5) Izolacja całkowitego RNA komórki, a w kolejnym etapie izolacji uzyskiwanie określonego RNA(cDNA) z zastosowaniem metody PCR [4].
Najczęściej stosowaną metodą jest izolacja całkowitego RNA komórki. Pozostałe metody stosowane są znacznie rzadziej. Rzadko wykorzystywana jest metoda oparta na izolacji RNA z polirybosomów.
Warunkiem uzyskania dobrej jakości preparatu RNA jest szybkie i skutecznie zahamowanie działania enzymów degradujących RNA tj. rybonukleaz. Rybonukleazy występują we wszystkich żywych komórkach i bardzo szybko niszczą cząsteczki RNA. Dlatego tez, do izolacji wymagane jest przygotowanie szkła i odczynników, które są wolne(pozbawione) rybonukleaz.
W związku z tym, wszystkie odczynniki powinny być przygotowywane na wodzie, do której wcześniej dodano dietylopirowęglan (DEPC). Najczęściej dodaje się go w ilości 1ml na 1 l wody. DEPC jest związkiem, który bardzo skutecznie hamuje działanie rybonukleaz. Jednakże należy pamiętać, że jest on także silnym mutagenem, dlatego należy pracować z nim pod wyciągiem i w rękawiczkach. Praca w rękawiczkach dotyczy także samego procesu izolacji RNA, ma to na celu uniknięcie zanieczyszczenia preparatów [4].
Izolacja RNA z tkanek (myszy) [4]Pobrać ok. 0,5 cm3 tkanki lub cały narząd myszy (np. śledzionę) i przenieść do probówki, w której znajduje się ciekły azot. Po wyparowaniu ciekłego azotu do tkanki dodać 200 μl BSS, 800 μl GTC, 100 μl 2M octanu sodu o pH=4.0, 1000 μl fenolu, oraz 200 μl mieszaniny chloroform: alkohol izoamylowy w stosunku 24:1. Tkankę homogenizować na lodzie przez około 1-3 minut. Po homogenizacji próbkę inkubować na lodzie przez 15 min. Fazę wodną oddzielić przez wirowanie w temp. 4˚C, przy 10 000 rpm, wirować przez 15 minut. Następnie dodać 1200 μl izopropanolu. Powstały osad RNA zebrać przez odwirowanie (4˚C, 10 000 rpm, 20 min). Po wirowaniu osad przemyć 2 razy 75% etanolem. Osad wysuszyć, a po wysuszeniu RNA zawiesić w 100 μl sterylnej wody traktowanej DEPC [4].
Izolacja całkowitego RNA z zastosowaniem TRIZOLU [4].Pobrany fragment tkanki (50-100 mg) homogenizować w TRIZOLU zawierającym fenol oraz izotiocyjanian guanidyny. Homogenat inkubować przez 5 minut w temperaturze 15-30 ˚C. Następnie dodać 0.2 ml chloroformu. Całość intensywnie mieszać i znów inkubować 3 minuty w temperaturze 15-30˚C. Rozdzielić fazy przez wirowanie w mikrowirówce (12 000rpm, 15 min, temp. 2-8˚C). Powstałą fazę wodną zebrać, dodać 0.5 ml izopropanolu i całość inkubować 10 minut w temperaturze 15-30˚C. Próbkę wirować w mikrowirówce (12 000rpm, 10 min, temp. 2-8˚C). Powstały supernatant usunąć.
Otrzymany osad RNA przemyć 1 ml 75% etanolu. Próbkę intensywnie wymieszać, a następnie zwirować (7500 rpm, 5 min., temp. 2-8˚C). Otrzymany osad RNA wysuszyć przez ok. 10 min, rozpuścić w wodzie lub 0.5% SDS. Całość wymieszać przez pipetowanie, a dalej inkubować 10 minut w temperaturze 55-60˚C. Otrzymany RNA przechowywać w temp. -70˚C [4].
W uproszczeniu, izolację całkowitego RNA można przedstawić w następujący sposób:
-krew (bądź inny materiał wyjściowy)
- zebranie komórek
- ekstrakcja RNA
- wirowanie
- wytrącanie RNA
- wirowanie
- rozpuszczanie RNA
- elektroforeza w żelu agarozowym i ocena RNA [4].
Izolacja RNA z linii komórkowychKomórki, z których ma być izolowany materiał genetyczny w postaci RNA, przepłukać 2x roztworem PBS. Następnie komórki zawiesić w odczynniku TRI® Reagent (Sigma), tj. na 10 000 000 komórek dodać 1 ml odczynnika. Dodać chloroform ( na 1 ml odczynnika TRI® Reagent dodać 0,2 ml chloroformu). Tak otrzymaną mieszaninę wytrząsać przez 10 sekund, a następnie zwirować przy 15 000 obr/min, 15 minut w 4˚C. Otrzymaną po wirowaniu fazę wodną przenieść do nowej probówki. Dodać 0,5 ml izopropanolu (0,5 ml izopropanolu na 1 ml odczynnika TRI® Reagent). Próbkę wstawić na 10 min do -20˚C (następuje wytrącanie RNA), a potem odwirować przez 20 min w 4˚C, przy prędkości 15000 obr./min.
Otrzymany osad RNA przemyć w 0,5 ml 70% etanolu, . Osad wysuszyć na powietrzu lub w suszarce próżniowej Speed Vac. Po suszeniu osad zawiesić w wodzie mQ. Tak otrzymaną próbkę RNA przechowywać w -20˚C [12].
Prowadzone w ostatnich latach intensywne badania potwierdziły, że wycinki utrwalane i rutynowo przeprowadzane do bloczków parafinowych, mogą być źródłem RNA o jakości wystarczającej do przeprowadzenie dalszych badań np. ekspresji genów [2].
Formalina jest 40% roztworem formaldehydu w wodzie, stosowanym w rozcieńczeniu w stosunku 1:9. Właściwości utrwalające formaliny są znane już od ponad 100 lat. Dlatego, też jest ona bardzo rozpowszechniona i powszechnie stosowana jako odczynnik utrwalający.
Formalina doskonale zachowuje morfologię tkanek, jednak tym samym wpływa niekorzystnie na strukturę kwasów nukleinowych. Formaldehyd reaguje z zasadami azotowymi występującymi w RNA, tworząc N-metylol (N-CH2OH) [2], [5].
Według przeprowadzonych badań, wszystkie zasady azotowe reagują z formaldehydem, jednakże najbardziej reaktywna jest adenina. Po inkubacji (16 godzinnej) w roztworze formaliny, w temp. 4˚C, wykazano, że prawie 40% cząsteczek adeniny w syntetycznym RNA zawiera addycyjny monometyl. Ponadto, addycja metylolu w komórkowym RNA, powoduje zakłócenia w czasie procesu odwrotnej transkrypcji oraz syntezy cDNA. Badania wykazały, że takie działanie można częściowo znieść, podczas podgrzewani próbek RNA w buforze TE (tj. Tris-EDTA). Poddanie RNA godzinnej inkubacji z 10 mM buforem TE, w temperaturze 70˚C powoduje, że częściowo usuwane są pochodne metylolu [16].
Doświadczenia, przeprowadzone przez Hamatani i wsp. wykazały, że chemiczne modyfikacje RNA pochodzące z tkanek, które utrwalane były w niezbuforowanej formalinie i przechowywane przez wiele lat, mogą zostać zniesione przez ogrzewanie próbek w buforze cytrynianowym (pH=3-6.5) [2].
Częściową poprawę jakości wyizolowanego RNA można uzyskać, stosując wspomnianą już inkubację próbek w buforze cytrynianowym. W przeprowadzonych doświadczeniach, najlepsze wyniki uzyskano stosując 30-60 minutową inkubację RNA w buforze cytrynianowym o pH=4.0. Najbardziej optymalna temperatura okazało się 70˚C [16].
Zastosowanie świeżych tkanek do izolacji materiału genetycznego, np.RNA , zapewnia stosunkowo dobrą jakość wyizolowanej próbki. Bardzo duże znaczenie dla jakości wyizolowanego RNA ma czas, jaki upłynął od momentu pozyskania danej tkanki, do momentu jej odpowiedniego zabezpieczenia. Według niektórych danych RNA w świeżych tkankach jest stabilne nawet do ok.6h ( w temperaturze pokojowej). Należy jednak mieć na uwadze, że niewłaściwe przechowywanie materiału do dalszych analiz (np. izolacji), niekorzystnie wpływa na ekspresję genów. Dlatego, też według doświadczeń wynika, że najbardziej wiarygodne dane pochodzą z próbek, w których pozyskana do badania tkanka została od razu przeniesiona na lód (ochrona przed degradacją przez RNAzy), lub do odpowiednich płynów, stosowanych komercyjnie do przechowywania tkanek [16].
Wycinki tkankowe, utrwalone w formalinie, a następnie w bloczkach parafinowych, są rutynowo wykorzystywane do diagnostyki histopatologicznej. Bardzo często stanowią też materiał do badań molekularnych, genomu, a także ekspresji genów, z użyciem różnych technik biologii molekularnej. Jednakże poważnym ograniczeniem jest wysoki stopień fragmentacji oraz chemicznych modyfikacji kwasów nukleinowych, które izolowane są z utrwalonych tkanek [2], [5].
Podczas izolacji materiału genetycznego z próbek, które wcześniej utrwalone były w formalinie, występuje kilka, znacznych ograniczeń. RNA, które pozyskiwane jest z tkanek utrwalonych w FFPE, charakteryzuje się niską jakością. Spowodowane to jest znacznym stopniem degradacji, która zachodzi jeszcze przed zakończeniem procesu utrwalania próbek w roztworze formaliny. Dodatkowo, już samo utrwalanie powoduje niekorzystne zmiany w próbce.
Między innymi, samo utrwalanie powoduje, że tworzone są wiązania krzyżowe pomiędzy cząsteczkami kwasów nukleinowych, a białkami. Ponadto, wprowadzane są modyfikacje kowalencyjne RNA. Ograniczenia te sprawiają, że utrudniona jest nie tylko izolacja RNA, ale także jego ocena (analiza) ilościowa i jakościowa [7], [8].
Izolacja RNA z tkanek utrwalonych w parafinie (FFPE, formalin-fixed, paraffin-embedded)Z próbek, z których ma być izolowane RNA, należy usunąć parafinę. Odbywa się to przez kolejne płukania próbek w ksylenie (4-8 krotnie), a następnie w alkoholu etylowym (99,8%, 3-4 krotnie). Badaną tkankę (po usunięciu parafiny) wysuszyć, homogenizować w 850 μl buforu denaturującego (tj. 4M izotiocyjanek guanidyny, 0.25 M cytrynian sodu, 0.5% sarkosyl, 0.1 M merkaptoetanol) z dodatkiem 250 μl roztworu proteinazy K (o stężeniu 20 mg/ml) zgodnie z Lehmann i Kreipe , a następnie inkubować w 55˚C przez noc (16-18h). Po inkubacji próbkę odwirować (14000g, 5 min, 4˚C). Po wirowaniu do otrzymanego supernatantu (200μl) dodać 1 ml odczynnika Trizol.
Kolejne etapy procedury przeprowadzić zgodnie z instrukcją producenta, tj. powstałą mieszaninę ekstrahować 0,2 ml chloroformu. Po ekstrakcji fazę wodną strącić przez dodanie 0,5 ml izopropanolu. Odwirować (12000g, 10 min, 4˚C), a otrzymany osad RNA płukać 75% alkoholem etylowym.Osad wysuszyć i rozpuścić w 100 μl buforu TE. RNA ponownie strącić przez noc w -70˚C stosując następującą mieszaninę: 220 μl alkoholu etylowego 99,8%, 10 μl 3M octanu sodowego. Całość odwirować (12000g, 10 minut, 4˚C). Osad wysuszyć i rozpuścić w wodzie (20-100μl) [7], [8].
Aktualnie dostępnych jest wiele gotowych zestawów do izolacji RNA. Jednakże bardzo często stosuje się tradycyjne protokoły izolacji, które opierają się m.in. na działaniu proteinazy K i tiocyjanianu guanidyny.
Podczas izolacji RNA z bloczków parafinowych metoda, w której stosuje się czynnik chaotropowy w postaci tiocyjanianu guanidyny- ustępuje metodom z zastosowaniem proteinazy K. A to dlatego, że dzięki enzymatycznemu trawieniu utrwalonej wcześniej tkanki, możliwe jest uzyskanie 10 razy więcej całkowitego RNA.
Istnieją również procedury, które łączą w sobie oba te czynniki. Godfrey i wsp. opisali 3-dniową inkubację z proteinazą K, w buforze zawierającym tiocyjanian guanidyny, a następni ekstrakcję za pomocą mieszaniny fenol: chloroform, z dodatkowym etapem oczyszczania [2], [5].
Enzymy zdolne do rozkładania kwasów nukleinowych , znane są od wielu lat. Enzymy swoiste dla kwasu deoksyrybonukleinowego nazywa się deoksyrybonukleazami (DNAzy), zaś te, które hydrolizują kwas rybonukleinowy, określane są jako rybonukleazy (RNAzy). W obrebie każdej z tych klas wyodrębnia się enzymy, które mają zdolność rozszczepiania wewnętrznych wiązań fosfodiestrowych. W wyniku rozszczepienia powstają końcówki 3’-hydroksylowe i 5’-fosforanowe, lub 5’-hydroksylowe i 3’-fosforanowe. Enzymy te określane są mianem endonukleaza. Niektóre z nukleaz zdolne są hydrolizować nukleotydy tylko wtedy, kiedy znajdują się one na końcu cząsteczki. Enzymami tymi są egzonukleazy [11], [13].
W badaniach wykazano, że niekorzystny wpływ na jakość RNA mogą mieć inhibitory RNAz, które dodawane są na etapie enzymatycznego trawienia tkanki. Pomimo tego, że RNAzy są enzymami bardzo stabilnymi, wysoce reaktywnymi i nie wymagającymi do działania kofaktorów, to jednak podczas utrwalania w formalinie są inaktywowane. Dodatkowo, użycie proteinazy K zapewnia ich całkowite zniszczenie [2].
Izolacja RNA z niewielkich bioptatów pobranych ze skóry człowieka i myszy [6].W 1,5 ml probówce umieścić 75-100 mg badanej próbki w probówkach zawierających żel blokujący, całość zwirować (12000xg, 30 s.). Pojedyncze bioptaty rozdrobnić na drobny proszek w schłodzonym moździerzu, standardowo używając ciekłego azotu.
Otrzymany proszek przenieść do 1,5 ml probówki, dodać 300 μl Qiazol (Qiagen). Homogenat zworteksować przez 15 s, a następnie wytrząsać przez 10 minut na Reax 2000 (Heidolph). Dodać 60 μl chloroformu, wirować przez 15 s, a po wirowaniu pozostawić w temperaturze pokojowej przez 3 minuty. Częściowo oddzieloną mieszaninę przenieść do wcześniej przygotowanych probówek, zawierających fazę żelu blokującego, i wirować przez 15 min, przy 12000xg. Otrzymaną fazę wodną przenieść do nowych 1,5 ml probówek. Otrzymane RNA oczyszczać przez wytrącanie kolumienek zgodnie z RNeasy MinElute Cleanup Handbook (wersja 2007). Dodatek D: Oczyszczanie RNA po lizie i homogenizacji z Qiazol Odczynnik do lizy (Qiagen). Na koniec procedury RNA eluować 14 μl wody (nuclease-free watre). Zwirować 1 minutę przy maksymalnych obrotach [6].
Według Bruning’a i wsp. przedstawiona wyżej metoda izolacji RNA z małych biopsji skóry człowieka i myszy, daje dostatecznej jakości RNA, które może być wykorzystywane do dalszych badań. Pomimo, iż metoda ta jest porównywalna pod względem jakości izolowanego materiału z innymi procedurami, to jednak jej zaletą jest to, że metoda ta jest łatwa do wdrożeni nawet w sytuacji braku specjalistycznego sprzętu laboratoryjnego.
Ponadto, Bruning i wsp. w swojej procedurze izolacji chwalą to, że powtarzalne wyniki ilości wyizolowanego RNA można uzyskać stosując bardzo małe skrawki skóry, wielkości 1.5 mm. Ma to ogromne znaczenie w przypadku wykonywania testów na skórze ludzi, ponieważ oznacza to, że materiał (skóra) do badań może być pozyskiwany ze znacznym zmniejszeniem dyskomfortu u osób badanych [6].
Izolacja RNA metodą AGPC-acid guanidinium/phenol/chloroform (według procedury opracowanej przez Chomczyńskiego i Sacchi) [1]
100 mg homogenizować w 2 ml roztworu zawierającego 4 M rodanek guanidyny i 25 mM cytrynian sodu (pH=7.0), 0,5% sarkosyl, 0.1 M 2-merkaptoetanol, całość inkubować 10-60 min w temperaturze pokojowej, od czasu do czasu mieszając. Następnie dodać 0.2 ml 2M octanu sodu (pH=4.0), 2 ml fenolu i 0.4 ml mieszaniny chloroform: alkohol izoamylowy (w stosunku 49:1), całość wymieszać. Przed wirowaniem (10 000xg, 20 min) mieszaninę ochłodzić na lodzie przez 15 min. Po wirowaniu i precypitacji etanolem otrzymaną próbkę RNA rozpuścić w 400 μl wody(RNase-free water) [1].
Zmodyfikowana metoda izolacji, opisana przez Shibata , Jackson’a i wsp., zawiera etap trawienia proteinazą K. W metodzie tej 100 mg próbki homogenizowano w 1 ml buforu trawiącego (tj. 200 mM Tris-HCl, 200 mM NaCl, 1.5 mM MgCl2, 2% SDS, pH=7.5) z 500 μg proteinazy K. Całość inkubowano w 45˚C przez 1 h. Lizat tkanki ekstrahować 1 ml płynnej mieszaniny fenol/chloroform/alkohol izoamylowy w stosunku 25:24:1, a następnie 1 ml chloroformu. Po precypitacji etanolem, dodać 400 μl wody (RNase-free water). W celu zapewnienia eliminacji genomowego DNA, próbkę inkubować w 37˚C przez 1 h z 1 U DNase I( na μg całkowitego RNA), a następnie inaktywować w 75˚C przez 5 minut [1].
Masuda i wsp. w swoim doświadczeniu testowali dwie metody izolacji RNA: metodę AGPC oraz metodę z proteinazą K. Jako materiał wykorzystywane były fragmenty wątroby utrwalony w 10% buforowanej formalinie(BF) i przechowywany przez okres 1 miesiąca po utrwaleniu w 4˚C. Plasterki tkanki o wadze ok. 100 mg używane były do izolacji RNA wymienionymi wyżej metodami.
W metodzie AGPC, nawet po intensywnej homogenizacji lub długotrwałej inkubacji w roztworze rodanku guanidyny do 1 h, małe fragmenty tkanki pozostawały widoczne. W przeciwieństwie do innego plasterka tkanki, który został całkowicie strawiony w buforze do lizy (zawierającym proteinazę K), przez inkubację w 45˚C przez 1h. Na podstawie powyższych wyników, uwzględniając różnicę w zdolności do rozpuszczania tkanki, należy zaznaczyć, że metoda z proteinazą K jest lepsza [1].
W przedstawionej zmodyfikowanej metodzie (z trawieniem proteinazą K) w doświadczeniu przeprowadzonym przez Masuda i wsp., wydajność wyizolowanego RNA była 10-razy większa w metodzie z trawieniem proteinazą K, niż w metodzie AGPC. W metodzie z proteinazą K nie stwierdzono różnic w ilości i integralności izolowanego RNA z tkanek świeżych i zamrożonych o podobnych rozmiarach [1].
RNA obecny w komórkach drożdżowych można wyekstrahować ze zhomogenizowanych komórek za pomocą metody, w której wykorzystuje się 2% roztwór soli sodowej siarczanu dodecylu (SDS). Otrzymany w ten sposób ekstrakt (RNA) charakteryzuje się niewielką zawartością DNA (tj. <0.5%), oraz białek, których jest < 2.0% [14].
Przedstawiona poniżej metoda izolacji RNA z komórek drożdżowych (Schmitt i wsp. ), pozwala na wyizolowanie wystarczającej ilości materiału w ok. 60 minut. W metodzie wykorzystano fenol oraz roztwór soli sodowej SDS [3].
Izolacja RNA z komórek drożdżowych Saccharomyces cerevisiae (mini-preparacja RNA z wykorzystaniem fenolu i SDS) [3].Komórki drożdży hodować w płynnym podłożu YPG ( 2% glukoza, 1% pepton, 1% ekstrakt drożdżowy). Pobrać 10 ml kultury, komórki zebrać przez odwirowanie, i zawiesić w 400 μl 50 mM octanu sodu o pH=5.3 oraz 10 mM EDTA (bufor AE).
Zawiesinę komórek przenieść do 1,5 ml probówki, dodać 40 μl 10% SDS. Całość zawiesiny zwirować. Następnie dodać świeży fenolu zrównoważony równą objętością buforu AE. Mieszaninę ponownie zwirować i inkubować w 65˚C przez 4 minuty, po inkubacji gwałtownie schłodzić na lodzie aż do wytrącenia się kryształów fenolu. Zwirować przez 2 minuty przy najwyższych obrotach, w celu oddzielenia fazy wodnej od fenolowej. Górną fazę (wodną) przenieść do nowej probówki, i ekstrahować ją mieszaniną fenol: chloroform w temperaturze pokojowej przez 5 minut. Do wyodrębnionej fazy wodnej dodać 40 μl 0.3M octanu sodu o pH=5.3. Następnie dodać 2,5 objętości alkoholu etylowego do powstałego osadu RNA. Osad przemyć 80% etanolem, wysuszyć i zawiesić w 20 μl sterylnej wody.
Przechowywać w -70˚C do momentu ponownego użycia próbki. Podczas izolacji podjąć powszechnie znane środki ostrożności w celu uniknięcia zanieczyszczenia RNA rybonukleazami [3].
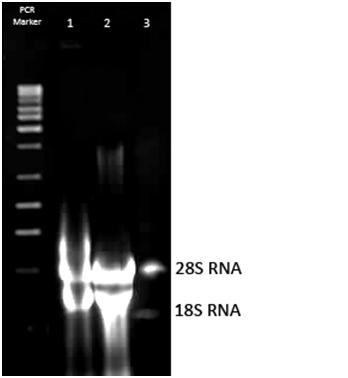
Zdjęcie: izolacja RNA z komórek drożdżowych Saccharomyces cerevisiae
Źródła własne, autor: L. KoperwasM.E. Schmitt, T.A. Brown i B.L. Trumpower [3] zastosowali skróconą metodę izolacji RNA z fenolem. Po izolacji otrzymano od 60μg do 300μg całkowitego RNA na 10 ml kultury, w zależności od szczepu z jakiego materiał genetyczny był izolowany [3]. Inna metoda izolacji RNA (z wykorzystaniem piasku szklanego-kulek szklanych) , uważana jest przez badaczy za żmudną w wykonaniu, a dodatkowo mało efektywną, gdyż izoluje się nią mało RNA [3].
Izolacja RNA z drożdżyW kolbie płaskodennej doprowadzić do wrzenia 10 mL 2% roztworu SDS. Następnie do wrzącego roztworu dodać 2 g rozdrobnionych świeżych drożdży. Całą zawartość ogrzewać przez ok. 3 minuty. Po ogrzewaniu, kolbę schłodzić strumieniem zimnej wody z kranu, następnie zawartość kolby przenieść do probówki wirówkowej i odwirować (10 min, 6000 rpm). Otrzymany po wirowaniu supernatant przenieść do kolbki płaskodennej, dodać 1 ml roztworu octanu potasu i 22 ml alkoholu etylowego. Mieszaninę (z wytrąconym RNA) pozostawić na łaźni lodowej przez okres 0,5h. Po upływie tego czasu próbkę odwirować (6 min, 6000 rpm). Roztwór znad osadu zdekantować, z kolei otrzymany osad RNA rozpuścić w 1 ml wody. Wyizolowane RNA z drożdży przechowywać w temperaturze 0°C do momentu dalszej analizy [14].
Izolacja RNA z komórek hodowanych in vitro [4].Komórki hodowane w naczyniu hodowlanym zebrać, przemyć PBS i zawiesić w 0,5 ml buforu A (tj.5 mM MgCl2, 100 mM NaCl, 50 mM HEPES pH=7.0). Dodać 0.5 ml buforu B (tj. bufor A, 1% Nonident P40, 0.2% DEPC) o temp. 0˚C. Całość dokładnie wymieszać, a następnie inkubować w temp. 0˚C przez 1 minutę. Otrzymane lizaty komórek wirować przez 5 minut, 2500 rpm, w temperaturze 4˚C (w celu oddzielenia jąder komórkowych).
Otrzymany po wirowaniu supernatant przenieść do nowej probówki Eppendorf. Do supernatantu dodać 0.6 ml mieszaniny fenol/CIAA w stosunku 1:1 (tj. fenol wysycony 10 mM Tris-HCl pH=7.5, 100 mM NaCl, 1 mM EDTA). Mieszaninę intensywnie wytrząsać prze 1 minutę. Następnie zwirować (20 min), zebrać powstały supernatant i ponownie wytrząsać z równą objętością fenol/CIAA. Po wirowaniu i zebraniu powstałej fazy wodnej, do próbki dodać 0.1 obj. 3 M octanu sodu o pH=5.2.
RNA wytracą przez dodanie 2,5 objętości etanolu. Odstawić, a po 3h zebrać osad RNA przez odwirowanie przez 30 minut przy 8000rpm. Otrzymany osad przemyć 80% etanolem, wysuszyć, a na koniec zawiesić w 100 μl sterylnej wody (traktowanej roztworem DEPC) [4].
Krew obwodowa jest to krew pochodząca z naczyń krwionośnych tj. z naczyń żylnych, włośniczek, bądź tętnic [15].
Izolacja RNA z limfocytów krwi obwodowej [4].10 ml krwi obwodowej pobrać na 100 μl 10% EDTA o pH=8.0. Krew rozcieńczyć (przez dodanie 10 ml BSS tj. roztwór A: D-glukoza 0.1%, 5x10-5M CaCl2 x 2H2O, 9,8x 10-6M MgCl2 x 6H2O, 5.4x10-3M KCl, 0.145M Tris-HCl o pH=7.6; roztwór B: 0.14M NaCl; roztwór końcowy: A:B=1:9). Całość nałożyć na 20 ml fikolu, zwirować w 18˚C przez 30 minut przy 1500 rpm. Po wirowaniu odrzucić surowicę. Zebrać 10 ml warstwy fikolu, która zawiera limfocyty.
Do otrzymanych limfocytów dodać 30 ml BSS, całość wymieszać, zwirować (18˚C, 10 min, 2500 rpm). Osad limfocytów zawiesić w 2 ml BSS, przenieść do nowej probówki typu Eppendorf, zwirować (15 sek, 10 000 rpm). Do powstałego osadu dodać 100 μl BSS i zawiesić komórki przez pipetowanie (5 razy). Do powstałej zawiesiny dodać 400 μl GTC (tj. 4M izotiocyjanian guanidyny, 25 mM cytrynian sodowy o pH=7.0, 0.5% sarkosyl, 0.1M 2-merkaptoetanol), przepipetować kilkakrotnie. Następnie dodać 50 μl 2M octanu sodu o pH=4.0, 500 μl fenolu (fenol wysycony H2O o pH=4.0), i 100 μl CIAA (tj. chloroform: alkohol izoamylowy, w stosunku 49:1). Całość dokładnie wymieszać przez pipetowanie (10 razy), i inkubować w temp. 0˚C przez 15 min. Oddzielić fazę wodną przez wirowanie (4˚C, 15 min, 10 000 rpm). Po wirowaniu dodać 600 μl izopropanolu i inkubować 2 h w temperaturze -20˚C.
Powstały osad RNA zebrać przez wirowanie (4˚C, 20 min, 10 000 rpm). Osad przemyć 2 razy 75% etanole. Po wysuszeniu próbkę RNA zawiesić w 20 μl wody (traktowanej DEPC) [4].
Autor: Lidia KoperwasLiteratura: [1]. Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K, 1999. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Research , 1999. Vol. 27, No. 22, 4436-4443.
[2]. Korga A, Wilkołaska K, Korobowicz E, 2007. Trudności w wykorzystaniu tkanek z archiwalnych bloczków parafinowych w badaniach ekspresji RNA. Postępy Higieny Medycyny Doświadczalnej (online), 2007; 61: 151-155.
[3] Schmitt M.E, Brown A.T, Trumpower B.L, 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic acid Research,Vol.18, No.10. s: 3091-3092
[4]. Słomski R, 2008. Analiza DNA, teoria I praktyka. Wydawnictwo Uniwersytetu Przyrodniczego w Poznaniu, Poznań 2008. s. 54-60.
[5]. Hamatani K, Eguchi H, Takahashi K, Koyama K, Mukai M, Ito R, Taga M, Yasui W, Nakachi K, 2006. Improved RT-PCR Amplification for Molecular Analyses with Long-term Preserved Formalin-fixed, Paraffin-embedded Tissue Specimens. Volume 54(7): 773–780, 2006. Journal of Histochemistry & Cytochemistry.
[6]. Bruning O, Rodenburg W, Radonic T, Zwinderman A.H, de Vries A, Breit T.M, de Jong M, 2011. RNA isolation for transcriptomics of human and mouse small skin biopsies. BMC Res Notes. 2011; 4: 438.
[7].[http://metlab.pl/Izolacja_RNA_z_tkanek_utrwalonych_w_parafinie_%28FFPE,_formalin-fixed,_paraffin-embedded%29_p26.html , dr Jadwiga MArchewka, 2009. Izolacja RNA z tkanek utrwalonych w parafilmie (FFPE, formalin-fixed, paraffin-embedded)]
[8]. Usarek E, Gajewska B, Kaźmierczak B, Kuźma M, Dziewulska D, Barańczyk-Kuźma A, 2005. A Study of Glutathione S-transferase pi Expression in Central Nervous System of Subjects with Amyotrophic Lateral Sclerosis Using RNA Extraction from Formalin –Fixed , Paraffin-Embedded Material. Neurochemical Research, Vol. 30, No. 8, August 2005 (_ 2005), pp. 1003–1007
DOI: 10.1007/s11064-005-6771-1.
[9]. Żak I, Kwasy nukleinowe. http://biochigen.slam.katowice.pl/podrecznik/19.pdf
[10]. Nalepa G,1995. Genetyka, wydanie II. Wyd. Helion, s.61-64
[11]. Murray R.K, Granner D.K, Mayes P.A, Rodwell V.W, 1995. Biochemia Harpera. Wydanie III. Wydawnictwo Lekarskie PZWL, s. 488, 739.
[12]. Piechota J, 2008. Izolacja RNA z linii komórkowych, http://metlab.pl/Izolacja_RNA_z_linii_komorkowych_p15.html
[13]. http://gmwh.republika.pl/index/enz.htm
[14]. [http://www.e-biotechnologia.pl/Artykuly/Izolacja-i-elektroforeza-RNA-drozdzy/]
[15]. [http://www.zdrowie.med.pl/bad_labor/badania/b_krwi.html]
[16]. http://metlab.pl/Stabilnosc_RNA_a_sposob_przechowywania_tkanek_p29.html










Recenzje