Pierwsze, nowoczesne badanie kliniczne zarejestrowano w 1948 roku. Badanie to polegało na porównaniu skuteczności ekspozycji słonecznej z terapią streptomycyną u chorych na gruźlicę.
Z kolei, podstawe zasady rejestracji badanych produktów generycznych zostały ustalone w1984 roku Stanach Zjednoczonych. W Europie podstawy reguł badan równoważności biologicznej w celu rejestracji produktów generycznych zostały zatwierdzone w 1991, po czym uzupełni ono je w 2001. Zazwyczaj mija kilkanaście lat od powstania koncepc ji oryginalnego produktu leczniczego do jego wejścia na rynek (rejestracji) [6].
Jak wiadomo, badania kliniczne stanowią główny etap procesu badawczo-rozwojowego danego produktu leczniczego ( leku). Każde badanie kliniczne poprzedzone jest tzw. badaniami podstawowymi oraz badaniami przedklinicznymi. Z kolei pomyślnie przeprowadzone badanie kliniczne zakończone jest procesem rejestracji i wprowadzeniem testowanego leku do obiegu [6].
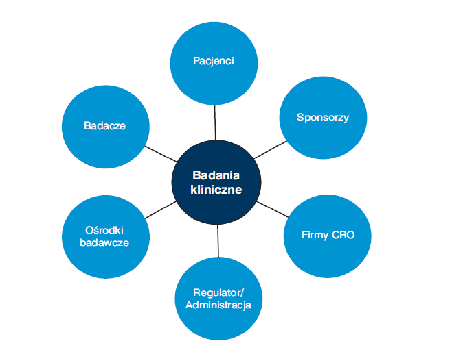
Uczestnicy badania klinicznego oraz podmioty zaangażowane W cały proces przeprowadzenia badania klinicznego zaangażowani są:
1) Sponsorzy
Do najczęściej odnotowywanych sponsorób badań klinicznych zalicza się firmy farmaceutyczne. Niektóre badania finansowane są także przez ośrodki badawcze lub instytucje akademickie. Tak, więc badania prowadzone są bezpośrednio albo przez firmy farmaceutyczne, albo zlecane są niezależnym firmom okrślanym jako CRO [7].
Sponsorem badania klinicznego jest:
- osoba fizyczna, osoba prawna albo jednostka organizacyjna nieposiadająca osobowości prawnej,
- odpowiedzialna za podjęcie, prowadzenie oraz finansowanie danego badania klinicznego,
- osoba, która ma siedzibę na terytorium jednego z państw członkowskich Unii Europejskiej lub państw członkowskich Europejskiego Porozumienia o Wolnym Handlu (EFTA).
W przypadku, gdy sponsor badania klinicznego nie ma siedziby na terytorium jednego z państw Europejskiego Obszaru Gospodarczego, może działać wyłącznie przez swojego prawnego przedstawiciela posiadającego siedzibę na tym terytorium.
Obowiązkiem Sponsora jest zapewnienie i kontrola jakości badania oraz standardowych procedur postępowania (SOP- Standard Operating Procedures). Prowadzenie badania, zbieranie, przechowywanie i raportowanie danych musi odbywać się zgodnie z protokołem badania, zasadami GCP oraz zgodnie z obowiązującymi przepisami. Sponsor musi także upewnić się, że we wszystkich ośrodkach będzie możliwy dostęp do danych i dokumentów źródłowych. Ma to na celu umożliwienie: monitorowania, audytów i inspekcji. Audytorzy przeprowadzający kontrolę badania muszą być niezależni i niezwiązani bezpośrednio z prowadzeniem badań klinicznych. Zadaniem sponsora jest powołanie odpowiednio wykwalifikowanego personelu medycznego, który będzie w stanie doradzać w kwestiach medycznych związanych z danym badaniem [7].
Zdjęcie: Uczestnicy badania klinicznego oraz podmioty zaangażowane w badanie [7].
2) CRO (czyli Contract Research Organization ) są to organizacje prowadzące badania kliniczne na zlecenie firm farmaceutycznych. Niezależnie wyspecjalizowane firmy CRO, skupione są wyłącznie na organizowaniu i nadzorowaniu badań klinicznych (co odróżnia je od firm farmaceutycznych). Firmy farmaceutyczne i CRO zatrudniają tzw. monitorów badań klinicznych (CRA- Clinical Research Associate). CRA stanowi wykwalifikowany personel, który odpowiedzialny jest za cały proces monitorowania badania klinicznego. Ponadto, CRO współpracują bezpośrednio z badaczami [7].
Sponsor może przekazać niektóre (lub wszystkie) swoje obowiązki i funkcje CRO, jednak ostateczna odpowiedzialność za jakość i zgodność danych zawsze pozostaje po stronie sponsora. Przekazanie obowiązków musi odbyć się drogą pisemną. CRO zobowiązana jest wprowadzić system zapewnienia jakości i jej kontroli w trakcie trwania badania. Aktualnie, w Polsce obecnych jest ok. 50 firm typu CRO: są to zarówno oddziały globalnych firm, jak i niewielkie (o lokalnym zasięgu).
3) Ośrodki badawcze – w zależności od rodzaju prowadzonych badań oraz obszaru terapeutycznego, badania kliniczne moga być przeprowadzane albo w warunkach ambulatoryjnych, albo w szpitalach. W przypadku, gdy badania prowadzone są w szpitalach, kluczową rolę w badaniach odgrywają dyrektorzy szpitali.
4) Badacze/Lekarze: mają bezpośredni kontakt z uczestnikami badania klinicznego (pacjentami). Badacze pracują albu indywidualnie albo z zespołem asystentów ( w zależności od zakresu proadzonych badań). Za wybór badacza oraz ośrodka badawczego odpowiada Sponsor.
Badaczem może być lekarz dentysta (jeżeli badanie kliniczne dotyczy stomatologii) albo lekarz weterynarii ( w przypadku badania klinicznego weterynaryjnego). Badacz musi posiadać prawo wykonywania zawodu na terytorium Rzeczypospolitej Polskiej, a także odpowiednio wysokie kwalifikacje zawodowe, wiedzę naukową i doświadczenie w pracy z pacjentami, które są niezbędne do prowadzonego badania klinicznego.Badacz jest osobą odpowiedzialną za prowadzenie badania klinicznego w danym ośrodku, w którym to badanie jest przeprowadzane. Jeżeli badanie kliniczne prowadzone jest przez zespół osób, badacz wyznaczony jest przez sponsora, za zgodą kierownika zakładu opieki zdrowotnej, w którym prowadzone jest badanie kliniczne. Badacz jest także kierownikiem zespołu, jest on odpowiedzialny za prowadzenie tego badania w danym ośrodku. W przypadku, kiedy badacz kieruje zespołem nazywamy go Głównym Badaczem(ang. Principal Investigator).Ponadto, badacz może przekazać (delegować) wybrane obowiązki innym osobom z prowadzonego zespołu badawczego, jednakże niezależnie od tego to badacz odpowiedzialny jest za badanie kliniczne w swoim ośrodku. Członkowie zespołu badawczego powinni mieć odpowiednie kwalifikacje (np. osoby podejmujące decyzje medyczne dotyczące uczestników muszą być lekarzami). Powinni także zostać zaznajomieni z informacjami dotyczącymi badania tj.: z protokołem, instrukcjami dotyczącymi badanego produktu, swoimi obowiązkami w badaniu.
Do obowiązków Badacza należy przechowywanie listy osób, które zaangażowane są w prowadzenie badania, a także udostępnianie jej monitorom, audytorom i inspektorom. Na liście tej powinny znaleźć się informacje do jakich obowiązków Główny Badacz upoważnił poszczególne osoby oraz wzory ich podpisów. Przy każdej zmianie osób zaangażowanych w badanie, konieczna jest aktualizacja listy. Co więcej, żadna procedura związana z badaniem klinicznym nie może być wykonywana przez osoby, których nie ma na tej liście.
5) Pacjenci: trafiają do badania klinicznego w wyniku dobrowolnego zgłoszenia. Ponadto, pacjent ma prowa do odmowy udziału w badaniu na każdym jego etapie, nie ponosząc z tego żadnych konsekwencji. Pacjenci poddają się badaniu zarówno w warunkach ambulatoryjnych jak i szpitalnych.
Co więcej, pacjent ma prawo do wszelkich informacji, które wiążą się z jego udziałem w danym badaniu. Jeżeli jakieś informacje są dla pacjenta niezrozumiałe, ma on prawo otrzymania szczegółowych wyjaśnień od lekarza oraz od personelu, który współpracuje z badaczem. Na każdem etapie prowadzonego badania klinicznego, uczestnik ma prawo do informacji o swoim stanie zdrowa, a także do ochrony swoich danych osobowych[8].
6) Regulatorzy rynku/Administracja: instytucje nadzorujące i komisje bioetyczne opiniuja pozytywne lub negatywne wyniki badań klinicznych, a także zatwierdzaja bądź odrzucają wniosek rozpoczęcia badania klinicznego. Rolą administracji jest też zapewnienie, że w trakcie trwania badania pzrestrzegane jest prawo oraz wymagania Międzynarodowej Konferencji ds. Harmonizacji (ICH) i Dobrej Praktyki Klinicznej (GCP)[7], [8].
Etyka w badaniach klinicznychPierwsze regulacje prawno-etyczne w badaniach klinicznych, nie dotyczyły bezpośrednio ochrony pacjenta, lecz miały na celu pomoc w kontroli „chemicznej czystości” produktów leczniczych i informacji o składzie danego produktu na etykietach. Do spełniania tych zadań powołano w USA w 1927 roku agencję rzadową:” Food and Drug Administration (FDA)”. Automatycznie, szybko okazało sie,że potrzeba jeszcze bardziej szczegółowych i wielopłaszczyznowych systemów kontroli i bezpieczeństwa nowych leków. Równolegle rosła świadomość potrzeby ochrony wśród samych pacjentów, co ciekawe nie tylko na obszarze jednego kraju czy regionu, ale w skali globalnej. Najbardziej dostrzegalnym „punktem zwrotnym” w etyce doświadczeń na ludziach była surowa ocena badan biomedycznych prowadzonych w obozach koncentracyjnych w czasie II wojny swiatowej, oraz powołanie do życia tzw. „Kodeksu Norymberskigo” (w 1947 r.). Dokument ten sformułowano w 10 punktach, i stał się on podstawą dla dalszych, systematycznych i coraz bardziej zaawansowanych prac nad etyką eksperymentu medycznego [1], [5].
W 1964 roku w Helsinkach podczas konferencji Światowego Stowarzyszenia Lekarskiego (World Medical Association –WMA) sformułowano tzw. Deklarację Helsińską, która określaął etyczne zasady prowadzenia badań medycznych z udziałem ludzi [3], [5].
Istotne treści Deklaracji Helsińskiej pozostają bez zmian od czasu jej sformułowania i dalszego aktualizowania i rozszerzania podczas kolejnych walnych zgromadzeń WMA w różnych częściach świata. Ogólnie najważneijsze części Deklaracji można ująć w 7 tezach:
„ 1. Każde badanie z udziałem ludzi powinno mieć podstawy naukowe i tam, gdzie to właściwe, powinno być poprzedzone badaniami laboratoryjnymi i badaniami z udziałem zwierzat.
2. Powinien istnieć protokół badania.
3. Protokół badania powinien być zatwierdzony przez niezależną (od badacza
i sponsora) Komisję Bioetyczną.
4. Uczestnik badania powinien być poinformowany o celu, metodzie, ryzyku i potencjalnych korzysciach badania.
5. Uczestnik badania powinien wyrazić zgodę na piśmie.
6. Badacz powinien posiadać odpowiednie kwalifikacje.
7 Badacz (i sponsor) jest odpowiedzialny za bezpieczeństwo pacjenta w badaniu” [1], [2].
Kolejnym kamieniem milowym w ochronie pacjenta uczestniczącego w badaniach klinicznych, było ustalenie Zasad Dobrej Praktyki Klinicznej (GCP). Zasady te zostały następnie uzupełnione przez Międzynarodową Konferencję ds. Harmonizacji (ICH). I tak od 1996 roku normy ICH GCP są przestrzegane na całym świecie i stanowią podstawę wszystkich zasad etycznych dotyczących prowadzenia badań klinicznych, które to w szczególny sposób mają za zadanie chronić człowieka poddanego badaniom eksperymentalnym [1], [5].
Autor: Lidia Koperwas
Literatura:
[1]. Dr Brusiło J., OFMConv., 2007. Badania kliniczne (eksperymenty medyczne) w kontekście wartości personalistycznych. Referat przedstawiony w ramach konferencji Chrzescijanskiego Forum Pracowników Nauki “Nauka-Etyka-Wiara” w Rogowie, 18-21 X 2007r.
[2]. Walter M., Miedzynarodowe uregulowania badan klinicznych – Deklaracja Helsinska, Zasady Dobrej Praktyki Badan Klinicznych (GCP), Miedzynarodowa Konferencja do spraw Harmonizacji (ICH) (w:) Badania kliniczne. Organizacja, nadzór i monitorowanie, ten_e (red.), Warszawa 2004, s. 49–50.
[3]. http://www.oil.org.pl/xml/oil/oil68/tematy/deklaracja_helsinska
[4].http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1Guideline.pdf
[5]. Brody B. A., The Ethics of Biomedical Research. An International Perspective, Oxford 1998, s. 213.
[6]. http://www.mtz-clinical.pl/downloads/projektowanie_badan_biorownowaznosci.pdf
[7].http://infarma.pl/fileadmin/badania_kliniczne_raport/Badania%20kliniczne%20w%20Polsce%202010.pdf
[8].http://www.badaniaklinicznewpolsce.pl/najwazniejsze-informacje-dla-uczestnika-badan/wazne-dla-uczestnikow-badan-klinicznych/bezpieczenstwo-i-prawa-uczestnika-badania-klinicznego/
Tagi:
badanie kliniczne,
sponsor,
badacz,
pacjent,
SOP,
CRO,
CRA,
ICH,
GCP,
komisja bioetyczna,
etyka badań klinicznych,
Deklaracja Helsińska,
Kodeks Norymberski,
WMA
wstecz
Podziel się ze znajomymi









Recenzje