
|

Zamknij X
|




Wstęp
Wirus zapalenia wątroby typu C (HCV) jest wirusem pandemicznym. W Polsce zakażonych tym wirusem jest prawie 700 000 osób, co stanowi około 1,5% populacji. Infekcje wirusem HCV mogą prowadzić do chronicznych chorób wątroby z wysokim ryzykiem rozwoju marskości wątroby oraz nowotworu wątrobowokomórkowego. Opracowanie skutecznej terapii przeciwko wirusowi zapalenia wątroby typu C jest trudne ze względu na trudności w opanowaniu metod hodowli wirusa i modelowych organizmów zwierzęcych. Dotychczas stosowana procedura leczenia zapalenia wątroby typu C zawiera kombinację interferonu-α (IFN-α) o zmienionej cząsteczce i przedłużonym działaniu z ribawiryną (RB). Oporność pacjentów z genotypem 1 HCV, najpowszechniej występującym na świecie, na terapię kombinowaną przy użyciu IFN-α i RB oraz wzrastająca liczba infekcji u ludzi młodych zmusza do poszukiwania bardziej efektywnych terapii przeciwko wirusowi zapalenia wątroby typu C [1].
Budowa i cykl replikacyjny wirusa HCV
Wirus zapalenia wątroby typu C jest otoczkowym wirusem ssRNA z rodziny Flaviviridae. Zbudowany jest on z lipidowej otoczki i białkowego kapsydu o strukturze ikosahedralnej, w skład którego wchodzi materiał genetyczny oraz białko rdzeniowe (Rys.1). Genom wirusa HCV stanowi jednoniciowy, liniowy RNA o długości około 9500 nukleotydów i dodatniej polarności. Białka wchodzące w skład wirusa HCV można podzielić na strukturalne oraz niestrukturalne. Do białek strukturalnych zalicza się: białko rdzeniowe C oraz białka otoczkowe E1 i E2, natomiast do białek niestrukturalnych: NS2, NS3, NS4A, NS4B, NS5a i NS5b. Białko rdzeniowe C bierze udział w tworzeniu wirionów potomnych przez wiązanie materiału genetycznego. Ponadto posiada ono zdolność hamowania apoptozy i replikacji wirusa HBV w przypadku współzakażenia wirusami zapalenia wątroby typu B i C. Białka otoczkowe E1 i E2 są glikoproteinami o różnych sekwencjach w zależności od genotypu wirusa HCV. W zakażonej komórce występują one w postaci kompleksów połączonych domenami hydrofobowymi z błonami siateczki śródplazmatycznej. Główną rolą glikoprotein E1i E2 jest udział w procesie wnikania wirionu do komórki. Białka niestrukturalne biorą udział w procesie replikacji HCV w komórce gospodarza [2].
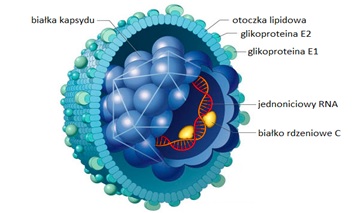
Rys.1. Budowa wirusa zapalenia wątroby typu C (HCV).
Cykl replikacyjny wirusa HCV rozpoczyna się w momencie przyłączenia wirionu do specyficznego receptora. Białkiem kompleksu receptorowego może być tetraspanina CD81, receptor rozpoznający zmodyfikowane przez utlenianie lub acetylację lipoproteiny o małej gęstości BI (SR-BI), receptor lipoprotein o małej gęstości (LDL) oraz cząsteczki adhezyjne DC-SIGN i L-SIGN. Wirus HCV przyłączony do kompleksu receptorowego jest następnie internalizowany a jego nukleokapsyd jest uwalniany do cytoplazmy. Po uwolnieniu materiału genetycznego wirusa z kapsydu bierze on udział w translacji poliproteiny oraz replikacji na terenie cytoplazmy. Replikacja i procesy potranslacyjne zachodzą w membranie utworzonej z wirusowych białek niestrukturalnych i białek gospodarza zwanych „kompleksem replikacyjnym". Następnie w retikulum endoplazmatycznym dochodzi do zamknięcia genomu wirusa w kapsyd. Nukleokapsydy są pokrywane otoczką lipidową i dojrzewają w aparacie Golgiego. Następnie nowo utworzone wiriony są uwalniane do przestrzeni zewnątrzkomórkowej na drodze egzocytozy [3].
Cele potencjalnych leków antywirusowych
Cykl replikacyjny wirusa HCV składa się z sześciu etapów:
Każdy z tych etapów może być potencjalnym celem leków antywirusowych (Tab.1) [4].
Tabela 1. Najważniejsze wirusowe i komórkowe czynniki wpływające na cykl replikacyjny wirusa HCV.
|
Etap cyklu |
Czynnik wirusowy |
Czynnik komórkowy |
|
Przyłączenie wirusa, wejście i fuzja |
Glikoproteina
powierzchniowa E1 Glikoproteina
powierzchniowa E2 |
Glukozoaminoglikany CD81 (cel
przeciwciał antyproliferacyjnych 1) Receptor
SR-BI Klaudyna-1 Białko DC-SIGN
lub CD209 Białko
L-SIGN lub DC209L Receptor
LDL Receptor
asialoglikoproteiny (ASGP-R) |
|
Translacja RNA HCV |
IRES Domena 1
5'UTR Otwarta
ramka odczytu HCV 3'UTR NS4A i NS4B |
Podjednostki
rybosomowe eIF2 i eIF3 tRNA Białko PTB hnRNP L Autoantygen
La Białko 2
wiążące Poli(rC) Białko 1
zasocjowane z NS1 |
|
Obróbka potranslacyjna |
Zależna od
Zn2+ metaloproteinaza NS2 NS3/4A
proteaza serynowa |
Peptydaza
sygnałowa Peptydaza
odcinająca sekwencję sygnałową |
|
Replikacja
HCV |
Zależna od
RNA polimeraza NS5B NS5A NS3
NTPaza/helikaza NS4B Region X
3'UTR Domena 1
5'UTR |
Fragmenty
błony lipidowej Ludzkie
białko hVAP-A Cyklofilina
B FBL2 Białko PTB miRNA
(mikroRNA) |
|
Złożenie oraz uwolnienie wirusa |
Białko
rdzenia Glikoproteiny
powierzchniowe E1 i E2 Genom RNA
HCV |
Membrany
ER Aparat
Golgiego |
Wirus zapalenia wątroby typu C wnika do wnętrza komórek gospodarza w złożonym i wieloetapowym procesie, który wymaga obecności wielu czynników. Przypuszczalnym receptorem dla wirusa HCV było szeroko rozpowszechnione komórkowe białko powierzchniowe CD81. Białko to należy do rodziny tetraspanin i jest zaangażowane w wiele procesów, takich jak adhezja, metastaza i ruchliwość komórek, aktywacja komórki i udział w transmisji sygnału. Białko CD81 posiada dwie pętle zaangażowane w wiązanie glikoproteiny E2 wirusa. Pomimo tego ekspresja jedynie CD81 nie jest wystarczająca do wniknięcia wirusa HCV do komórki. Prowadzi to do wniosku, iż białko CD81 może funkcjonować jako koreceptor, czyli cząsteczka pomocna podczas infekcji i może spełniać funkcję po związaniu wirionu z innym receptorem [4].
Kolejnym białkiem receptorowym dla wirusa HCV jest ludzki receptor rozpoznający zmodyfikowane przez utlenianie lub acetylację lipoproteiny o małej gęstości klasy B typu I (SR-BI). SR-BI jest glikoproteiną posiadającą dwie domeny cytoplazmatyczne przedzielone przez dużą domenę zewnątrzkomórkową uczestniczącą w komórkowym metabolizmie tłuszczów. SR-BI przyłącza się do rejomu hiperprzemiennego HVR1 glikoproteiny E2 wirusa. Udział SR-BI w wejściu wirusa HCV do komórki został potwierdzony w receptorowych testach kompetycyjnych przy użyciu przeciwciał poliklonalnych anty-SR-BI. Sugeruje to na udział SR-BI w wniknięciu wirusa HCV do komórki. Jednak wiele ludzkich linii komórkowych z koekspresją białek CD81 i SR-BI jest nie-wrażliwych na infekcję wirusa HCV, co wskazuje na istnienie innych molekuł wymaganych do wniknięcia wirusa HCV [4,5].
Jako potencjalne receptory dla wirusa HCV badano także lektyny typu C (zależne od Ca2+), takie jak L-SIGN, DC-SIGN i receptor asialoglikoproteiny (ASGP-R). ASGP-R występuje w błonie komórkowej hepatocytu. Natomiast L-SIGN i DC-SIGN nie występują w hepatocytach, przez co nie mogą być receptorami HCV. Wykazano jednak, że ich rola w wniknięciu wirusa HCV do komórki może być związana z transportem HCV do hepatocytów. Kolejnym potencjalnym receptorem dla wirusa HCV jest receptor LDL, który jest zdolny do regulacji internalizacji HCV poprzez wiązanie zasocjowanych z wirionem cząstek LDL [4,6].
Przeprowadzono również badania nad udziałem klaudyny-1 (CLDN1) w wnikaniu wirusa HCV do komórki. CLDN1 jest składnikiem struktur międzykomórkowych połączeń ścisłych, głównych komponentów kompleksów adhezji międzykomórkowej, które tworzą intramembranę przepuszczalną dla jednych i nieprzepuszczalną dla innych cząstek. Białko to ulega ekspresji we wszystkich tkankach nabłonka, a przede wszystkim w wątrobie. Klaudyna-1 składa się z 211 aminokwasów i posiada dwie pętle zewnątrzkomórkowe, cztery segmenty transmembranowe i trzy domeny wewnątrzkomórkowe. Domena pierwszej pętli zewnątrzkomorkowej prawdopodobnie bierze udział w wejściu wirusa HCV do komórki. Przeprowadzone badania funkcjonalne CLDN1 wykazują, że białko to działa w fazie infekcji już po związaniu wirusa HCV z CD81 prawdopodobnie po związaniu z SR-BI. Sugeruje to na udział innych czynników w wniknięciu wirusa HCV do komórki [4,7]. Prowadzono również badania nad inną komponentą kompleksu adhezji międzykomórkowej, która może mieć udział w inicjacji infekcji wirusem HCV. Okludyna jest białkiem posiadającym cztery regiony transmembranowe, dwie pętle zewnątrzkomórkowe oraz N- i C-końcowy region cytoplazmatyczny. Bierze ona udział w adhezji międzykomórkowej w rejonie zamykającym przestrzeń międzykomórkową i kotwiczącym kompleksy adhezji międzykomórkowej do cytoszkieletu. Liczne badania wskazują na akumulację okludyny w retikulum endoplazmatycznym i jej kolokalizację z glikoproteiną E2 wirusa. Wykazano, że hamowanie ekspresji CLDN1i okludyny prowadzi do zahamowania wejścia wirusa HCV do komórki [4,8].
Glikozoaminoglikany (GAG) są to powierzchniowe polisacharydy mogące pełnić rolę pierwotnych receptorów o niskim powinowactwie i brać udział we wstępnej interakcji wirusa HCV z powierzchnią komórki gospodarza przed związaniem z receptorami wysokospecyficznymi. Większość glikozoaminoglikanów zawiera dodatkowo grupę siarczanową. Prawdopodobnie siarczan heparanu (HS) jest odpowiedzialny za interakcję pomiędzy wirusem HCV a komórką gospodarza, ułatwiając jego orientację. Jednak badania z udziałem cząstek wirusa HCV posiadającego na swej powierzchni heterodimery E1-E2 nie doprowadziły do wniknięcia wirusa do komórki. Sugeruje to, że wiązanie siarczanu heparanu (HS) nie jest dostępne dla dimeru E1-E2 lub kontakt wirusa HCV z komórkowymi GAG regulowany jest przez lipoproteiny zasocjowane z cząstkami wirusa [4,9].
Wyniki przeprowadzonych badań wykazują, że wirus HCV wymaga kooperacji wielu receptorów w celu efektywnego wniknięcia do komórki gospodarza. Fakt ten potwierdza, że blokada pojedynczego receptora tylko częściowo hamuje infekcję. Neutralizacja ludzką surowicą lub przeciwciałami monoklonalnymi anty-E2 redukuje aktywność wirusa HCV w stosunku do receptora glikoproteiny E2, ale nie znosi jej całkowicie. Preparaty obniżające ekspresję receptorów wirusa HCV na powierzchni hepatocytów, mogą być wykorzystane do blokowania wniknięcia wirusa do komórki gospodarza [10].
Po związaniu wirusa HCV z receptorami na powierzchni komórki gospodarza następuje fuzja otoczki lipidowej wirusa z błoną komórkową gospodarza. Proces ten jest skoordynowany w czasie i przestrzeni oraz wymaga licznych zmian konformacyjnych w białkach błony lipidowej, uwalnianych przez czynniki komórkowe. Wirus HCV wnika do wnętrza komórki gospodarza poprzez endocytozę, natomiast fuzja zachodzi we wczesnych endosomach. Kwaśne pH endosomów inicjuje proces fuzji prawdopodobnie poprzez zmiany konformacyjne w białkach otoczki wirusa. Ekspozycja wirionów związanych z powierzchnią komórek gospodarza do kwaśnego środowiska, po której następuje powrót do pH obojętnego, nie wpływa na infekcyjność wirusa HCV. Prowadzi to do wniosku, że białka otoczki wirusa HCV wymagają dodatkowych wyzwalaczy, aby uzyskać wrażliwość w środowisku kwaśnym. Po fuzji otoczki wirusa z membraną endosomalną genom wirusowy jest uwalniany do cytoplazmy [11].
Glikoproteiny E1 i E2 otoczki wirusa HCV posiadają aktywność czaperonową i uczestniczą w procesie fuzji. Różne regiony tych glikoprotein mogą współdziałać ze sobą w czasie fuzji. Wykazano, że mutacje w domenach transmembranowych białek E1 i E2 wpływają na właściwości fuzyjne glikoprotein otoczki wirusa HCV poprzez reorganizację oligomerów białek fuzyjnych. Prawdopodobnie cząstki lub krótkie peptydy współzawodniczące z tymi regionami glikoprotein E1 i E2, które regulują indukowaną przez niskie pH rearanżację struktury powierzchni wirusa HCV powinny posiadać aktywność antywirusową. Opierając się na tej teorii skonstruowano inhibitory dla wirusowych białek fuzyjnych dla wirusa HIV-1. Cyjanowiryna-N wchodzi w interakcję z glikoproteinami otoczki wirusa HCV i blokuje interakcję pomiędzy białkami E2 i CD81. Cząsteczki specyficznie wiążące się z glikoproteinami otoczki wirusa HCV mogą mieć znaczenie w rozwoju terapii antywirusowej [4,12].
Dekapsydacja wirionu prowadzi do uwolnienia genomowego RNA wirusa do cytoplazmy komórki. Uwolniony RNA razem z nowo zsyntetyzowaną cząsteczką RNA służy jako mRNA do syntezy poliproteiny HCV. Najbardziej konserwatywnym fragmentem genomu wirusa HCV jest region 5'UTR. Zawiera on 341 nukleotydów zlokalizowanych przed kodonem inicjatorowym otwartej ramki odczytu (ORF). Region 5'UTR zawiera 4 domeny, oznaczane I do IV, posiadające elementy strukturalne typu wypętleń, pseudohelis czy pseudowęzła. Domeny II, III i IV razem z pierwszymi 12 do 30 nukleotydami regionu kodującego stanowią miejsce wiązania rybosomu (IRES). Domena I 5'UTR nie należy do IRES, ale pełni rolę regulatorową. IRES jest regionem regulatorowym procesu translacji materiału genetycznego wirusa HCV. Pośredniczy on w niezależnej od struktury czapeczki (5'-metyloguanozyny) inicjacji translacji poliproteiny HCV przez rekrutację białek wirusowych i komórkowych, w tym czynników inicjujących (elF) 2 i 3. W pierwszym etapie translacji IRES formuje stabilny kompleks preinicjatorowy przez związanie podjednostki rybosomowej 40S. Następnie podjednostka 40S przyłącza eIF3, eIF2, GTP oraz inicjatorowy tRNA, w celu uformowania kompleksu preinicjującego 48S. W kompleksie tym tRNA znajduje się w pozycji P podjednostki 40S rybosomu. Kolejnym etapem jest hydroliza GTP, podczas której eIF2 uwalnia inicjatorowy tRNA i oddysocjowuje od kompleksu. Hydroliza następnej cząsteczki GTP z udziałem eIF5B pozwala na asocjację podjednostki 60S i uformowanie funkcjonalnego rybosomu 80S, który inicjuje syntezę białek wirusowych [4,13].
Inicjacja translacji może zostać zahamowana na etapie wejścia RNA do rybosomów przez blokowanie regionu 5'UTR RNA wirusowego IRES. VGX-410C jest związkiem hamującym formowanie kompleksu inicjującego i translację poprzez blokowanie wiązania IRES i podjednostki 40S z eIF3 [14].
Translacja białek wirusa HCV może być zahamowana przez antysensowne oligonukleotydy DNA lub RNA, których sekwencja jest komplementarna do wirusowego mRNA. Oligonukleotydy skierowane na 5'UTR hamują jego ekspresję w systemach kultur komórkowych. Zastosowanie syntetycznego oligomeru AVI-4065 hamuje translacje białek wirusa HCV zarówno in vitro jak i na modelowych organizmach zwierzęcych [14].
Różne typy rybozymów są zdolne do specyficznej inhibicji translacji poliproteiny HCV in vitro. Rybozymy są cząsteczkami RNA rozpoznającymi specyficzne sekwencje nukleotydowe i katalizującymi cięcie cząsteczki RNA. Niezmodyfikowane rybozymy są degradowane przez nukleazy komórkowe, ale modyfikacja chemiczna może zwiększać ich stabilność [15].
Istotną role w dojrzewaniu wirusa odgrywa rozszczepienie wirusowych poliprotein. W tym procesie uczestniczą zarówno enzymy pochodzenia wirusowego jak i komórkowego. Do cięcia proteolitycznego białek strukturalnych wirusa HCV wymagane są dwie peptydazy komórek gospodarza, peptydaza sygnałowa i peptydaza odcinająca sekwencje peptydu sygnałowego oraz dwie peptydazy wirusowe biorące udział w obróbce białek NS2 i NS3/4A wirusa HCV [4,16].
NS2 jest wirusowym białkiem transmembranowym i wraz z domeną białka NS3 stanowi proteinazę NS2-3. Proteinaza ta jest metaloproteinazą zależną od cynku rozcinającą białka NS2 i NS3. Białko NS2 traci swoją aktywność proteolityczną po odcięciu NS3. Natomiast NS3 jest wirusowym białkiem zawierającym domenę proteazy serynowej i domenę NTP-azową/helikazową. Białko NS4A jest kofaktorem aktywności proteolitycznej NS3 o działaniu stabilizującym i lokalizującym enzym w membranie ER. Proteaza serynowa NS3/4A katalizuje hydrolizę poliproteiny HCV i rozcina połączenia pomiędzy NS3/NS4A, NS4A/NS4B, NS4B/NS5A oraz NS5A/NS5B [4,16].
Białko prekursorowe generowane przez translację materiału genetycznego wirusa HCV jest kierowane do membrany ER w celu translokacji ektodomeny E1 do wnętrza ER. Proces ten jest regulowany przez sekwencję sygnałową zlokalizowaną pomiędzy rdzeniem i sekwencją E1. Peptydaza sygnałowa komórki gospodarza rozcina wiązanie pomiędzy glikoproteinami E1 i E2 wewnątrz ER. Dodatkowa sygnalaza rozcina fragment C-końcowy glikoproteiny E2 oraz wiązanie pomiędzy p7 i NS2. Białka E1 i E2 podlegają następnie procesowi dojrzewania, na który składają się glikozylacja przy końcu N, fałdowanie i formowanie heterodimerów E1E2. Dimery E1E2 uczestniczą w formowaniu cząsteczki wirusa. Proteinaza NS2-3 rozcina wiązanie pomiędzy NS2 i NS3. Dochodzi do uformowania kompleksu NS3 z jego kofaktorem NS4A, który katalizuje cięcie połączeń pomiędzy NS3-NS4A, NS4A-NS4B, NS4B-NS5A i NS5A-NS5B [4,16].
Zaprojektowano i zsyntetyzowano wysoce selektywne inhibitory wirusowej proteinazy NS3/4A. BILN-2061 to makrocykliczny inhibitor HCV NS3/4A. Powoduje on znaczne i szybkie obniżenie poziomu wirusowego RNA w stosunku do HCV genotypu 1 i w mniejszym stopniu u pacjentów zakażonych genotypem 2 oraz 3. Badania BILN-2061 wykazują, że zahamowanie aktywności proteinazy NS3/4A skutkuje inhibicją replikacji wirusa HCV. Innym inhibitorem proteinazy serynowej NS3/4A jest Telaprevir. Preparat ten jest silnym kowalencyjnym, przyswajalnym doustnie inhibitorem peptydowym. Telaprevir wykazuje aktywność antywirusową w stosunku do dzikich szczepów wirusa HCV i niektórych opornych na działanie BILN-2061 [4,17].
W celu zahamowania funkcji proteinazy NS3/4A zsyntetyzowano cząstkę acylotimocznika ACH-806/GS-9132, która hamuje wiązanie NS4A do NS3 i zatrzymuje dojrzewanie poliproteiny przez uniemożliwienie formowania aktywnego kompleksu proteinazy. ACH-806/GS-9132 posiada wysoką aktywność in vitro przeciwko genotypowi 1 wirusa HCV. Wyniki te wykazują, że przyłączenie NS4A do NS3 może być celem nowych inhibitorów proteinazy NS3/4A [4,18].
Domena transmembranowa białka NS5B jest odpowiedzialna za potranslacyjną lokalizację funkcjonalnej domeny tego białka do cytoplazmatycznej strony ER. Interakcje pomiędzy subdomenami białka NS5B powodują, że centrum katalityczne jest całkowicie otoczone, co zapewnia syntezę dodatniej (+) i ujemnej (-) nici RNA HCV. NS5B wiąże białko cyklofilinę B, komórkową peptydylo-prolylo cis-trans izomerazą, która reguluje replikację wirusa HCV poprzez modulację zdolności wiązania RNA przez zależną od RNA polimerazę RNA (RdRp) [4,19].
NS5A jest ufosforylowaną Zn2+-metaloproteiną, która odgrywa istotną rolę w replikacji wirusa HCV. N-koniec białka NS5A jest odpowiedzialny za lokalizację tego białka w membranie perinuklearnej, a także za utworzenie kompleksu replikacyjnego. Prawdopodobnie poziom fosforylacji NS5A odgrywa ważną rolę w wirusowym cyklu życiowym poprzez regulację przejścia z replikacji do składania wirusa. Funkcją formy hiperfosforylowanej jest utrzymanie kompleksu replikacyjnego w formie niezdolnej do przejścia w etap składania. Ponadto wykazano, że białko NS5A jest zdolne do interakcji z białkiem NS5B oraz posiada aktywność modulującą RdRp [20].
Domenę NTPazy/helikazy tworzą C-końcowe aminokwasy białka NS3. Białko to jest odpowiedzialne za stymulowaną RNA aktywność NTPazy, wiązanie RNA oraz rozplatanie regionów RNA o ekstensywnej strukturze drugorzędowej poprzez wiązanie, rozplatanie i hydrolizę trójfosforanów nukleozydów [21].
NS4B jest integralnym białkiem membranowym zlokalizowanym w ER lub membranie pochodzącej z ER. Zawiera on 4 domeny transmembranowe i N-końcowy fragment heliakalny, które są odpowiedzialne za asocjację membrany. Choresterol i kwasy tłuszczowe zawarte w membranie ER wpływają na jej płynność, co może modulować replikację wirusa HCV. Ponadto białko NS4B jest odpowiedzialne za kotwiczenie kompleksu replikacyjnego [22].
Replikacja wirusa HCV jest semikonserwatywna i dwuetapowa. Obydwa etapy procesu replikacji są katalizowane przez NS5B RdRp. Podczas pierwszego etapu, replikacji wirusa, dodatnio spolaryzowana nić RNA służy jako matryca do syntezy przejściowej nici ujemnej. W drugim etapie ujemna nić RNA jest używana jako matryca do wytworzenia wielu nici o dodatniej polarności, które następnie zostaną wykorzystane do syntezy poliprotein, syntezy nowych pośredników replikacji lub pakowania do nowych cząstek wirusowych. RdRp HCV jest zdolna do syntezy RNA de novo w specjalnych warunkach doświadczalnych [23].
Kompleksowość procesu replikacji i liczba składników biorących w niej udział oferuje wiele potencjalnych celów dla leków antywirusowych. Inhibitory zależnej od RNA polimerazy RNA należą do dwóch grup: inhibitory nukleozydowo/nukleotydowe, skierowane na centrum katalityczne enzymu oraz inhibitory nienukleozydowe skierowane na miejsca allosteryczne RdRp. Wyróżniamy cztery miejsca allosteryczne w strukturze polimerazy RNA. Obecnie trwają poszukiwania różnych cząsteczek, które mogą wybiórczo wiązać się z nimi i hamować aktywność RdRp. Miejsce A znajdujące się na szczycie domeny kciuka RdRp, z którym wiążą się indole i benzymidazole; miejsce B w domenie kciuka, które jest celem pochodnych fenyloalaniny, tiofenów, dwuhydroksypironów i pyranoindoli; miejsce C w domenie dłoni, z którym wiążą się benzotiadiazyny, acylopirolidyny, sulfonamidów proliny lub pochodnych kwasu akrylowego oraz miejsce D będące celem benzofuranów [4,24].
Inhibitory nukleozydowe należą do kategorii 4'-azydocytydyny lub 2'-C-metylocytydyny. Ostatnio zsyntetyzowany MK-0608 należy do kategorii 2'-C-metylocytydyny. Preparat ten gwałtownie obniża poziom RNA HCV. Natomiast NM-283 lub Valopicitabina jest prolekiem NM-107, analogiem nukleozydu 2'-C-metylocytydyny. Kolejnym lekiem wprowadzonym do I fazy badań klinicznych jest nienukleozydowy inhibitor RdRp, A-837093. W ostatnich latach syntetyzuje się coraz więcej analogów nukleozydów oraz prowadzone są nowe terapie przy użyciu analogów, które nie wymagają fosforylacji do pełnej aktywności wewnątrzkomórkowej [4,25].
Cyklosporyna A jest silnym inhibitorem cyklofiliny B. Mechanizm jej działania polega na antagonizacji efektu cyklofiliny B na replikację wirusa HCV. Otrzymano także syntetyczne analogi nieimmunosupresyjne cyklosporyny B. Jeden z nich - DEBIO-025 jest silniejszym inhibitorem replikacji wirusa HCV od cyklosporyny A. Efekt antywirusowy DEBIO-025 jest niezależny od genotypu HCV [4,26].
Prowadzone są także liczne badania nad wytworzeniem inhibitorów helikazy HCV. Inhibitory NTPazy/helikazy mogą działać według kilku mechanizmów. Pierwszy z nich polega na inhibicji aktywności NTPazy przez blokadę kompetytywną miejsca wiązania NTP lub za pomocą mechanizmu allosterycznego. Drugi prowadzi do zahamowania przekazywania energii i zmian konformacyjnych poprzez immobilizację regionu „przełącznika". Kolejny mechanizm polega na inhibicji kompetytywnej wiązania RNA. Natomiast ostatni z mechanizmów hamuje rozplatanie przez blokadę sferyczną translokacji NTPazy/helikazy wzdłuż łańcucha polinukleotydowego. Zsyntetyzowano selektywny inhibitor helikazy HCV imitujący nukleotyd QU-663. Wykazano, że aptamery RNA wiążą się z domeną NS3 helikazy i kampetytywnie hamują jej aktywność enzymatyczną [4,27].
Białko rdzenia wirusa HCV jest zasadowym polipeptydem wiążącym RNA, które formuje wirusowy kapsyd. Zawiera ono 3 domeny: hydrofilową domenę N-końcową (domena D1), hydrofobową domenę C-końcową (domena D2) oraz aminokwasy końcowe. Domena D1 jest odpowiedzialna za wiązanie RNA i lokalizację jądrową, domena D2 odpowiada za asocjację białka rdzenia z membranami ER, zewnętrznymi membranami mitochondriów i kropelkami lipidów. Natomiast końcowe aminokwasy pełnią rolę peptydu sygnałowego dla glikoproteiny E1 [4,28].
Białka rdzenia HCV mogą asocjować i formować struktury podobne do kapsydu. Prawdopodobnie formowanie cząsteczki wirusa jest inicjowane przez interakcję białka rdzenia z genomowym RNA. Interakcja rdzeń-RNA może odgrywać istotną rolę w przełączeniu z replikacji do pakowania wirusa. Zasocjowanie glikoprotein E1 i E2 z membranami ER sugeruje, że składanie wirusa może zachodzić w ER. Natomiast białka strukturalne zaobserwowano zarówno w ER, jak i aparacie Golgiego, co sugeruje, że obydwie struktury uczestniczą w procesie dojrzewania. Nowo wytworzone cząstki wirusa opuszczają komórkę gospodarza poprzez konstytutywny szlak sekrecyjny [29].
Wykazano, że iminocukry są zdolne do przekraczania membrany komórkowej oraz gromadzenia się w ER. Związki te mogą kompetytywnie hamować α-glukozydazę znajdującą się w ER, zmieniać glikozylację białek płaszcza oraz wpływać na składanie wirusa HCV. UT-231B jest pochodną iminocukru posiadającą zdolność inhibicji α-glukozydazy [4,29].
Podsumowanie:
Cykl replikacyjny wirusa HCV posiada wiele potencjalnych celów dla leków antywirusowych. Zahamowany może zostać każdy z etapów tego cyklu. Istnieje jednak wiele trudnych do przekroczenia barier w procesie wytwarzania inhibitorów wirusa HCV. Duży problem stanowi toksyczność w badaniach przedklinicznych in vitro uzyskana na modelach zwierzęcych lub we wczesnych etapach prób klilicznych oraz niska efektywność in vivo, która często kontrastuje z dobrymi wynikami antywirusowymi uzyskanymi na modelach przedklinicznych. Utrudnienie stanowi także odporność charakteryzowana przez szybką selekcję wariantów wirusa posiadających substytucje aminokwasowe zasocjowane z brakiem wrażliwości na lek. Niemniej jednak, ogromna liczba potencjalnych terapii antywirusowych stwarza możliwość zastąpienia stosowanej obecnie terapii IFN-α i ribawiryną. Jednakże wprowadzenie do lecznictwa leków hamujących replikację wirusa HCV, wymaga prowadzenia dalszych badań przed- oraz klinicznych na populacjach pacjentów.
Słowa kluczowe: wirus zapalenia wątroby typu C (HCV), cykl replikacyjny, leki antywirusowe.
Literatura:
25 maja 2018 roku zacznie obowiązywać Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 z dnia 27 kwietnia 2016 r (RODO). Potrzebujemy Twojej zgody na przetwarzanie Twoich danych osobowych przechowywanych w plikach cookies. Poniżej znajdziesz pełny zakres informacji na ten temat.
Zgadzam się na przechowywanie na urządzeniu, z którego korzystam tzw. plików cookies oraz na przetwarzanie moich danych osobowych pozostawianych w czasie korzystania przeze mnie ze strony internetowej Laboratoria.net w celach marketingowych, w tym na profilowanie i w celach analitycznych.
Administratorami Twoich danych będziemy my: Portal Laboratoria.net z siedzibą w Krakowie (Grupa INTS ul. Czerwone Maki 55/25 30-392 Kraków).
Chodzi o dane osobowe, które są zbierane w ramach korzystania przez Ciebie z naszych usług w tym zapisywanych w plikach cookies.
Przetwarzamy te dane w celach opisanych w polityce prywatności, między innymi aby:
dopasować treści stron i ich tematykę, w tym tematykę ukazujących się tam materiałów do Twoich zainteresowań,
dokonywać pomiarów, które pozwalają nam udoskonalać nasze usługi i sprawić, że będą maksymalnie odpowiadać Twoim potrzebom,
pokazywać Ci reklamy dopasowane do Twoich potrzeb i zainteresowań.
Zgodnie z obowiązującym prawem Twoje dane możemy przekazywać podmiotom przetwarzającym je na nasze zlecenie, np. agencjom marketingowym, podwykonawcom naszych usług oraz podmiotom uprawnionym do uzyskania danych na podstawie obowiązującego prawa np. sądom lub organom ścigania – oczywiście tylko gdy wystąpią z żądaniem w oparciu o stosowną podstawę prawną.
Masz między innymi prawo do żądania dostępu do danych, sprostowania, usunięcia lub ograniczenia ich przetwarzania. Możesz także wycofać zgodę na przetwarzanie danych osobowych, zgłosić sprzeciw oraz skorzystać z innych praw.
Każde przetwarzanie Twoich danych musi być oparte na właściwej, zgodnej z obowiązującymi przepisami, podstawie prawnej. Podstawą prawną przetwarzania Twoich danych w celu świadczenia usług, w tym dopasowywania ich do Twoich zainteresowań, analizowania ich i udoskonalania oraz zapewniania ich bezpieczeństwa jest niezbędność do wykonania umów o ich świadczenie (tymi umowami są zazwyczaj regulaminy lub podobne dokumenty dostępne w usługach, z których korzystasz). Taką podstawą prawną dla pomiarów statystycznych i marketingu własnego administratorów jest tzw. uzasadniony interes administratora. Przetwarzanie Twoich danych w celach marketingowych podmiotów trzecich będzie odbywać się na podstawie Twojej dobrowolnej zgody.
Dlatego też proszę zaznacz przycisk "zgadzam się" jeżeli zgadzasz się na przetwarzanie Twoich danych osobowych zbieranych w ramach korzystania przez ze mnie z portalu *Laboratoria.net, udostępnianych zarówno w wersji "desktop", jak i "mobile", w tym także zbieranych w tzw. plikach cookies. Wyrażenie zgody jest dobrowolne i możesz ją w dowolnym momencie wycofać.
Więcej w naszej POLITYCE PRYWATNOŚCI




Recenzje