Zastosowanie do oznaczania kwasów nukleinowych metod chemicznych, opiera się na określeniu ilości jednego ze składników wchodzących w skład kwasu nukleinowego. Tak więc wśród badanych i oznaczanych składników znajdują się albo kwas fosforowy, albo ryboza ( w przypadku RNA lub deoksyryboza- gdy pracujemy na DNA) oraz zasady azotowe. Dokładność z jaką uda się określić zawartość danego składnika zależy głównie od czułości zastosowanej metody analitycznej, a także od zawartości procentowej poszczególnych składników w kwasach nukleinowych [9]. Słowa kluczowe: absorbancja, stężenie kwasów nukleinowych, żel agarozowy, rozdział elektroforetyczny, pomiar spektrofotometryczny, czipowa technika Bioanalyzer 2100
Słowa kluczowe: absorbancja, stężenie kwasów nukleinowych, żel agarozowy, rozdział elektroforetyczny, pomiar spektrofotometryczny, czipowa technika Bioanalyzer 2100Po wyizolowaniu kwasów nukleinowych tj. DNA lub RNA, jednym z pierwszych etapów jaki się wykonuje jest określenie ich czystości i stężenia. Zarówno stężenie DNA jak i RNA można określić dwoma sposobami. Pierwszym z nich może być pomiar absorpcji światła UV przez wyizolowane materiały. Z kolei druga metoda opiera się na wybarwieniu preparatu bromkiem etydyny, następnie wykonaniu elektroforezy w żelu agarozowym, a na końcu porównanie fluorescencji preparatu z preparatem o znanym stężeniu [8].
Preparaty do pomiaru absorbancji muszą być wcześniej rozcieńczone (najczęściej 100 x H2O lub buforem TE). Oznaczenie wykonuje się za pomocą pomiary fluorescencji (spektrofotometrem), przy czym należy pamiętać, aby do kalibracji spektrofotometru użyć tego samego buforu, w którym wcześniej rozcieńczono preparaty. Następnie po kalibracji spektrofotometru odczytuje się punktowo wartość absorpcji przy 260, 280 i 320 nm, bądź też można wykonać widmo pomiarów w zakresie od 200 do 350 nm. Wśród najczęściej stosowanych znajdują się spektrofotometry typu NanoDrop. Charakteryzują się one znacznie skróconą drogą optyczną , jednocześnie przy zachowaniu bardzo dużej dokładności pomiarów. Urządzenia tego typu są bardzo przydatne do pomiarów stężenia DNA i RNA, w momencie kiedy dysponuje się unikatowym materiałem wyjściowym (niewielkie jego ilości) [8].
Przyjęło się, że absorbancja przy 260 nm jest równa 1 dla dwuniciowego DNA (dsDNA) o stężeniu 50μg/ml, dla jednoniciowego DNA o stężeniu 33 μg/ml, oraz dla RNA o stężeniu równym 40 μg/ml. Różna złożoność struktury DNA i RNA powoduje różnice w absorbancji. W związku z tym jednoniciowe kwasy nukleinowe absorbują więcej światła [8].
Mierząc stężenie kwasów nukleinowych przy długości fali równej 260 i 280 nm, możliwe jest jednoczesne określenie ich czystości. Otóż, stosunek wartości absorpcji A260 do A280 świadczy o zanieczyszczeniu preparatów białkami. Wartość tego współczynnika między 1.8-2.0 oznacza, że wyizolowane preparaty są wystarczająco oczyszczone, z kolei wartość równa 1.5 świadczy o tym, że w danym preparacie kwasów nukleinowych (DNA lub RNA) jest 50% białka, w związku z czym preparat taki wymaga dodatkowego oczyszczenia [8].
Należy mieć na uwadze fakt, że na ustalenie stężenia DNA lub RNA ma wpływ obecność w preparacie innych substancji, dlatego też zaleca się, aby wykonywać odczyt stężeń przy kilku długościach fali.
Tabela: Substancje absorbujące i charakterystyczne dla nich długości fali [8].
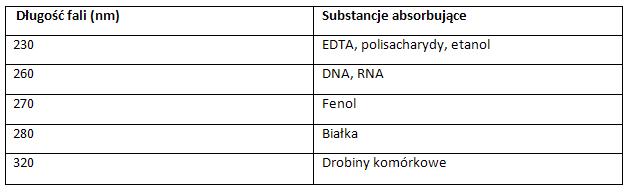
Inna często stosowaną metodą oceny ilościowej preparatów kwasów nukleinowych jest porównanie po elektroforezie w żelu agarozowym z preparatem o znanym stężeniu. Metoda ta zaliczana jest do metod z wyboru- w przypadku gdy dysponujemy bardzo małymi ilościami objętościami preparatów DNA. Dzięki zastosowaniu rozdziału DNA w żelu agarozowym, możliwe jest dodatkowo określenie jakości preparatu [8].
W celu sprawdzenia zarówno jakości, jak i ilości preparatu DNA należy przygotować 0,8% żel agarozowy, zawierający 0,5 μ/ml bromku etydyny w buforze 1 x TBE (tj. 10 x TBE: 0,89 M Tris, 0,89 M kwas borowy, 0,02 M EDTA, pH=8.0), następnie nakłada się 0,5; 1,0; oraz 5,0 μg preparatu o znanym stężeniu, oraz preparaty do oszacowania (w buforze obciążającym). Następnie po przeprowadzonej elektroforezie, ilość i jakość preparatów ocenia się na trans iluminatorze, który emituje światło o długości 312 nm. Jeżeli w porównaniu do markera, wielość wyizolowanego DNA migruje w postaci wartego prążka o wielkości ponad 50 kpz, bez widocznych smug, o oznacza to, że wyizolowane DNA jest wysokocząsteczkowe [4], [8].
Metody pomiaru jakości RNACząsteczka RNA odgrywa kluczową rolę w przekazywaniu informacji zakodowanych w genomie (DNA) do różnych form białka. Po ekstrakcji RNA z komórek za pomocą różnych metod, możliwy jest bezpośredni pomiar aktywności komórkowej, wykorzystując w tym celu różne techniki pomiaru ekspresji genów. Tak więc wśród tych metod wyróżnia się m.in. Real-Time PCR, czy mikromacierze DNA (są to aktualnie najczęściej stosowane techniki) [2].
RNA jest termodynamicznie stabilną cząsteczką, która jest jednak bardzo szybko trawiona przez niemal wszechobecne enzymy RNazy. W celu oceny stopnia degradacji RNA przez enzymy stosuje się metody elektroforetyczne, które polegają na separowaniu próbek według wielkości występujących w nich fragmentów kwasów nukleinowych. Zazwyczaj integralność RNA oceniana jest za pomocą elektroforezy w żelu agarozowym, wybarwionym bromkiem etydyny, co powoduje uwidocznienie w żelu pewnych pasmowych wzorów (tzw. prążków). Zazwyczaj na zdjęciach żeli widoczne są dwa pasma obejmujące 28S i 18S rybosomalne RNA (rRNA). RNA jest uznawane za wysokiej jakości, gdy stosunek pasm (prążków) 28S : 18S jest równy około 2.0 i wyżej. Takie podejście opiera się na ludzkiej interpretacji obrazów żelu, w związku z czym ocena taka jest subiektywna , a także trudno porównywalna między np. dwoma różnymi laboratoriami, a ponadto otrzymane dane rozdziału elektroforetycznego nie mogą być przetwarzane cyfrowo [2]. Szczególnie długie fragmenty mRNA do 10 kb są bardzo wrażliwe na degradację [3].
Ekstrakcja i procedury oczyszczania całkowitego RNA muszą spełniać następujące kryteria:
• nie zawierać białka (absorbancji 260 nm/280 nm);
• wolne od genomowego DNA;
• powinny być niezdegradowane (stosunek 28S do 18S powinien być mniej więcej między 1,8 i 2,0, z
małą ilością krótkich fragmentów);
• wolne od wszelkich substancji takich jak Mg lub Mn2 +
• wolne od nukleaz dla długotrwałego przechowywania [3].
Praca z RNA niskiej jakości może poważnie zagrozić wynikom doświadczalnym i dalszym wnioskom, które są często pracochłonne, czasochłonne i wysoce drogie. Korzystanie z nienaruszonego RNA jest kluczowym elementem dla skutecznego stosowania nowoczesnych molekularnych metod biologicznych, jak QRT-PCR lub analiza mikromacierzy. Obecnie, na rynku dostępne są systemy do automatycznej elektroforezy kapilarnej, które są na dobrej drodze, by stać się standardem w ocenie jakości RNA. Generowane profile dostarczają informacji na temat stężenia RNA, pozwalają na wizualną kontrolę integralności RNA, i generują zbliżone proporcje między masą podjednostek rybosomu [3].
W przypadku oceny jakości wyizolowanego RNA , można zastosować 3 metody, tj. : ocena gęstości optycznej w żelu agarozowym, ocena spektrofotometryczna preparatu, oraz jedną z nowszych metodą tzw. czipową technikę Bioanalyzer 2100 [5].
Ocena gęstości optycznej w żelu agarozowym jest jedną z najstarszych metod stosowanych w laboratoriach. W metodzie tej oznacza się stosunek podjednostek 28S do 18S po rozdziale próbki RNA w żelu agarozowym z dodatkiem bromku etydyny. Elektroforezę prowadzi się w 1 lub 1,5% żelu agarozowym, w warunkach denaturujących. Rozdział prowadzi się przez ok. 2 godziny przy napięciu 70V. W wyniku rozdziału otrzymuje się 2 dobrze widoczne frakcje RNA tj. 28S i 18S. Intensywność świecenia frakcji 28S powinna być dwa razy intensywniejsza niż frakcji 18S. Zachowanie tej zależności świadczy o niewystąpieniu degradacji wyizolowanego materiału. Podczas rozdziału RNA na żelu może być widoczna także fakcja mRNA (w postaci smugi), a także migrująca jako pierwsza frakcja tRNA oraz niskocząsteczkowe RNA [5], [6], [8].
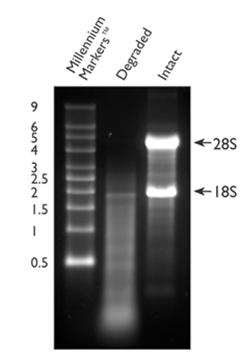
Zdjęcie: Frakcje RNA: 28S i 18S, http://www.biocompare.com/Articles/ApplicationNote/388/Is-Your-RNA-Intact-Methods-To-Check-RNA-Integrity.html [6]
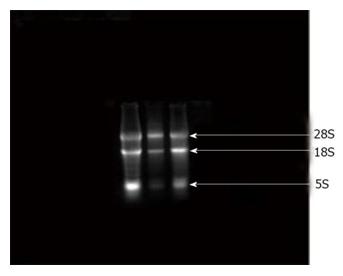 Zdjęcie:
Zdjęcie: Trzy frakcje prawidłowo wyizolowanego RNA (28S, 18S i 5S RNA), http://www.wjgnet.com/1948-5182/full/v2/i6/WJH-2-233-g001.htm [7].
Pomiar spektrofotometryczny wyizolowanego RNA prowadzi się przy różnych długościach fali (podobnie jak i DNA). Pomiar absorbancji przy 240 nm, pozwala na określenie tła i ewentualnych kontaminacji (podobnie jak pomiar przy 320 nm). Długość 260 nm jest specyficzna dla kwasów nukleinowych, z kolei pomiar przy 280 nm jest długością charakterystyczną dla białek.
Na podstawie wykreślonego OD 260 nm odczytujemy ilość RNA natomiast stosunek OD 260/280 mówi nam o jakości wyizolowanego preparatu. Stosunek długości fali 260/240 nm lub 260/320 nm wskazuje na czystość wyizolowanej próbki. W przypadku, gdy stosunek pomiarów przy długości fal 260/280nm jest większy niż 1,8 to wynik jest akceptowany jako RNA o dobrej jakości. Obecność genomowego DNA w próbce podczas pomiaru OD 260 może prowadzić do przeszacowania realnej ilości RNA [5].
Bardziej specyficznym spektroforetycznym pomiarem jest użycie odpowiednich sond znakujących np. RNA RiboGreen dye. Użycie sond pozwala wykryć już 1ng RNA/ml. W porównaniu do pomiaru spektrofotometrycznego, metoda ta jest bardziej specyficzna i nie oznacza ewentualnych zanieczyszczeń próbki białkami i DNA [5].
Czipowa technika Bioanalyzer 2100W 1999 r. do rozdzielania DNA, RNA i próbek białka został wprowadzony Agilent 2100 bioanalyzer. Od tego czasu jest on główną techniką analizy próbek RNA. Bioanalyzer jest automatycznym bioanalitycznym urządzeniem, wykorzystującym technologię Microfluidics, który zapewnia eletroforetyczną separację w automatyczny i powtarzalny sposób. Niewielkie ilości próbek RNA są rozdzielane w kanałach w zależności od ich masy cząsteczkowej, a następnie wywołana detekcja fluorescencyjna jest wykrywane przez laser. Rezultatem są dane przedstawione jako elektroforogram, gdzie ilość mierzonej fluorescencji koreluje z ilością RNA o danym rozmiarze [2]. Ponieważ dane są produkowane w formacie cyfrowym, mogą być z łatwością przetwarzane w celu umożliwienia dodatkowych obliczeń na podstawie uzyskanych danych surowych [2].
Czipowa technika Bioanalyzer 2100 służy do określania integralności RNA jako liczby od 1-10 w zależności od jakości badanej próbki materiału genetycznego. Jest to jedna nowszych metod, wykonywana z wykorzystaniem urządzenia Agilent 2100. Podstawę działania tego specyficznego urządzenia stanowi połączenie elektroforezy mikrokapilarnej , mikropłynów, mikroczipów i detekcji fluorescencyjnej. Pomiar wykonywany jest automatycznie, a jedną z zalet tego urządzenia jest fakt, że do analizy wymagane są niewielkie ilości badanego materiału. Analizie można przeprowadzić dysponując już 200 pg RNA.
Dzięki specyficznemu programowi, możliwe jest określenie całościowego RNA w systemie numerowym od 1 do 10, gdzie 1 wskazuje, że RNA jest najbardziej zdegradowane, zaś 10, że jest ono najlepszej jakości. Wynik analizy próbki otrzymuje się w postaci wykresu [4], [5].
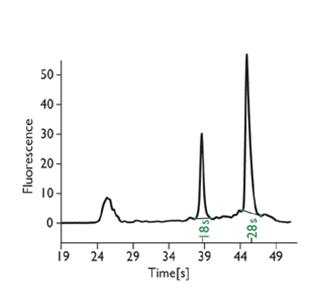
Zdjęcie: Wykres rozdziału elektroforetycznego próbki RNA, piki 18S i 28S http://www.biocompare.com/Articles/ApplicationNote/388/Is-Your-RNA-Intact-Methods-To-Check-RNA-Integrity.html [6].
Proces degradacji RNA jest tylko częściowo zrozumiały, ponieważ jest to zależne od rodzaju RNazy, która jest obecna i często jest w połączeniu z procesami fragmentacji. Ponadto, jakość RNA w danym eksperymencie może różnić się w znacznym stopniu od ekstrakcji RNA w innym eksperymencie i musi być pod stałym nadzorem. Przy użyciu precyzyjnej aparatury analitycznej, takiej jak Agilent 2100 bioanalyzer, eksperci są w stanie odróżniać próbki RNA o różnej jakości [2].
Aktualnie za jedną z najbardziej dokładnych metod służących do pomiaru stężenia DNA uważa się ilościowy PCR w czasie rzeczywistym tj. RT PCR (ang. Real-Time PCR). Ten rodzaj PCR dostarcza szczególnie cennych informacji odnośnie ilości oraz jakości amplifikowanego DNA. W porównaniu do innych metod, RT PCR charakteryzuje się specyficznością gatunkową. Ponadto metoda nie daje fałszywie zawyżonych wyników, które spowodowane są obecnością domieszki obcych kwasów nukleinowych , bądź innych związków wywołujących interferencję [1].
Elektroforeza kapilarnaDzięki postępowi technologicznemu możliwe jest określenie wielkości i stężenia wyizolowanych kwasów nukleinowych znacznie szybciej i precyzyjniej niż dotychczas, a to wszystko za sprawą automatyzacji systemów rozdziału elektroforetycznego biocząsteczek. Rozwój ten zapewniło pojawienie się technik opartych na elektroforezie kapilarnej w tzw. mikrochipach (ang. chip capillary electrophoresis). Tak jak już wspomniano, jednym z pierwszych komercyjnie dostępnych systemów wykorzystujących technologię rozdziału kwasów nukleinowych oparta na chipach jest 2100 Bioanalyzer (Agilent). Do analizy różnych cząsteczek (o różnej wielkości) zaprojektowano odpowiednie odczynniki oraz płytki chipowe [1].
W wyniku przeprowadzonych badań, wielu badaczy stwierdziło, że metoda ta sprawdza się nie gorzej od wcześniej wykorzystywanych metod. Jedną z wielu zalet elektroforezy chipowej jest szybka analiza nawet wielu próbek , oraz niższa ocena analizy jednej próbki w porównaniu do komercyjnie stosowanych testów w identyfikacji osobniczej. Aparat 2100 Bioanalyzer pozwala na analizę 12 próbek w ciągu pół godziny [1]. Wykorzystując ten typ aparatu możliwa jest analiza fragmentów DNA nie przekraczających kilkunastu tysięcy par zasad [1]. Do oznaczania wielkości i stężenia fragmentów dwuniciowego DNA ( w zakresie od 1000 do 12000 pz) zaprojektowano zestaw Agilent DNA 12000 LabChip. Wielkość cząsteczek DNA uzyskanych podczas izolacji klasyczną metodą organiczna, mieści się w zakresie 10000-15000pz, zaś za pomocą zestawu DNA IQ System w zakresie 60-10000pz [1].
W doświadczeniu przeprowadzonym przez Gorzkiewicz i wsp. przeprowadzono ocenę przydatności systemu do elektroforezy chipowe, zastosowanej do wstępnej analizy DNA badanego dla potrzeb sądowych. W wyniku przeprowadzonych badań (Gorzkiewicz i wsp.), badacze wywnioskowali, że system 2100 Bioanalyzer w połączeniu z zestawem Agilent DNA LabChip z powodzeniem może być wykorzystywany do analizy ilościowej genomowego DNA, jednakże pomiar ten dotyczy tylko określonych zakresów stężeń [1].
Według przeprowadzonych badań (Gorzkiewicz i wsp.) połączenie obydwóch systemów nie nadaje się do dokładnej oceny stężenia ekstraktów, zarówno tych, które zawierają zdegradowane DNA, jak i tych zawierających bardzo duże stężenia materiału genetycznego (DNA) [1].
Ponadto, system 2100 Bioanalyzer może być wykorzystany do ogólnej oceny wielkości fragmentów DNA (w próbkach zawierających stosunkowo duże stężenie DNA o dobrej jakości). W wyniku doświadczeń stwierdzono, że system 2100 Bioanalyzer niestety nie znajduje większego zastosowania do oceny DNA pochodzącego ze śladów biologicznych (ze względu na trudności w przewidzeniu stężenia i jakości DNA) [1].
Autor: Lidia KoperwasLiteratura:[1]. Gorzkiewicz M, Duleba A, Rychlicka E, Woźniak M, Grzybowski T, Śliwka K, 2010. Evaluation Of The Agilent 2100 Bioanalyzer As A Tool For DNA Analysis In Forensic Genetics, Problems of Forensic Sciences 2010, vol. LXXXI, 91-100.
[2]. Schroeder A, Mueller O, Stocker S , Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T, 2006. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Molecular Biology 2006, 7:3 doi:10.1186/1471-2199-7-3
[3]. Fleige S, Pfaffl W.M., 2006. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine 27 (2006) 126–139
[4]. Ugaz V.M, 2007. Electrophoresis in Microfluidic Systems. Artie McFerrin Department of Chemical Engineering Texas A&M University College Station, TX 77843, USA.
[5].http://metlab.pl/Oznaczanie_jakosci_RNA_bardzo_wazny_krok_w_badaniach_molekularnych_p38.html
[6]. http://www.biocompare.com/Articles/ApplicationNote/388/Is-Your-RNA-Intact-Methods-To-Check-RNA-Integrity.html
[7]. http://www.wjgnet.com/1948-5182/full/v2/i6/WJH-2-233-g001.htm
[8]. Słomski R, 2008. Analiza DNA, teoria I praktyka. Wydawnictwo Uniwersytetu Przyrodniczego w Poznaniu, Poznań 2008. S.61-64
[9]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s. 347-349











Recenzje