 W chromatografii fazy odwróconej podstawą jest oddziaływanie hydrofobowych obszarów cząsteczek białkowych oraz ligandu, które trwale są umocowane na powierzchni odpowiedniego nośnika. Hydrofobowe ligandy stosowane w technice RPC są znacznie gęściej rozmieszczone na powierzchni nośnika, niż w złożach do chromatografii oddziaływań hydrofobowych (HIC) [1], [12].
W chromatografii fazy odwróconej podstawą jest oddziaływanie hydrofobowych obszarów cząsteczek białkowych oraz ligandu, które trwale są umocowane na powierzchni odpowiedniego nośnika. Hydrofobowe ligandy stosowane w technice RPC są znacznie gęściej rozmieszczone na powierzchni nośnika, niż w złożach do chromatografii oddziaływań hydrofobowych (HIC) [1], [12].
W technice RPC zazwyczaj wykorzystuje się ligandy alkilowe, których łańcuchy zbudowane są z 4 do 18 atomów węgla (C4 – C18). W konsekwencji zastosowania właśnie takich ligandów dochodzi do bardzo silnego i wielopunktowego wiązania białek. Do elucji białek wymagane jest zastosowanie niepolarnych rozpuszczalników, co niestety może być przyczyną zajścia nieodwracalnych zmian aktywności biologicznej dużych cząsteczek białkowych. Tak więc, chromatografia fazy odwróconej ma ograniczone zastosowanie, a mianowicie nie używa się jej do preparatywnej chromatografii enzymów i receptorów białkowych, z kolei metoda ta znalazła bardzo szerokie zastosowanie praktyczne w przypadku małocząsteczkowych związków organicznych (zarówno w skali analitycznej jak i w preparatywnej), [1], [12].
Złoża do chromatografii fazy odwróconejZłoża do RPC wykonywane są w odmianach przeznaczonych dla chromatografii HPLC (wysokociśnieniowej). Kolumny wypełnione tymi złożami mogą procować albo w technice izokratycznej, albo w gradientowej.
Technika izokratyczna polega na dostarczaniu przez system pomp solwentu, który nie ulega zmianie w czasie rozdziału. W technice gradientowej skład solwentu ulega zmianie w trakcie trwania rozdziału, przy czym każda zmiana jest bardzo dokładnie kontrolowana. W zależności od zastosowanej techniki (gradientowej czy izokratycznej) otrzymuje się różne wyniki separacji makromolekuł, na co wpływ mają odmienne procesy zachodzące w obrębie kolumny chromatograficznej w trakcie rozdziału. Metodę gradientową można podzielić na dwa etapy:
- w pierwszym zachodzi sorpcja makromolekuł na hydrofobowym ligandzie, a warunki sorpcji dobierane są w taki sposób, by zapewnić najlepszą ekspozycję hydrofobowych obszarów na powierzchni separowanych makromolekuł. W przypadku białek kolumna zazwyczaj zrównoważona jest za pomocą zakwaszonej wody. W dalszym etapie przemywa się kolumnę za pomocą solwentu startowego (co ma na celu usunięcie nieswoiście zaadsorbowanych cząsteczek).
- w drugim etapie metody, system pomp dostarcza eluent charakteryzujący sie narastającym stężeniem niepolarnego rozpuszczalnika organicznego. W tym celu można zastować m.in. : metanolu lub acetonitryl. Na tym etapie, faza ruchoma staje się bardziej atrakcyjnym srodowiskiem dla zaadsorbowanych na unieruchomionym ligandzie cząsteczek – dochodzi do zmiany preferowanej przez separowane cząsteczki fazy : z fazy nieruchomej na ruchomą. To właśnie ten proces określono : „odwracaniem faz” [12].
Selektywność eluowania cząsteczek zależy od szybkości narastania gradientu stężenia solwentu niepolarnego: im wolniej zachodzi proces formowania gradientu, tym selektywność rozdziału cząsteczek jest większa. Automatycznie dochodzi do wydłużenia czasu trwania procesu separacji. W celu poprawy selektywności (bez zmiany czasu trwania całego rozdziału) stosuje się metodę z tzw. gradientem nieliniowym bądź metodę ze zróżnicowaną szybkością zmian gradientu w różnych odcinkach czasu. Do zalet metod gradientowej należy możliwość łatwego uzyskania informacji o stężeniu niepolarnego solwentu (co jest niezbędne do elucji z kolumny interesującej nas cząsteczki) [12].
Gdy znamy stężenie niepolarnego solwentu możemy tak ustawić program chromatografii, by w możliwie krótkim czasie uzyskać stężenie będące nieco poniżej stężenia niezbędnego do elucji, a w dalszym etapie procesu bardzo powoli dochodzić do warunków wymywania danej cząsteczki z kolumny [12].
Zalety chromatografii fazy odwróconej(RPC):
- w technice gradientowej nie ma ograniczeń co do objętości próbki nanoszonej na kolumnę, a także co do stopnia rozcieńczeń danej próbki
- możliwość znacznego skrócenia czasu rozdziału (praca w technice izokratycznej)
- technika chromatografii fazy odwróconej pozwala na odsalanie i zatężanie próbki
- technika charakteryzuje się wysoką rozdzielczością i selektywnością [2].
Wady RPC:
- stosowanie bardzo kosztownych solwentów charakteryzujących się najwyższą czystością
- niektóre stosowane solwenty moga degradować biomolekuły
- ograniczony wybór solwentów [2].
Rozdział chromatograficzny krótkich peptydów (wyodrębnianych z hydrolizatu fibrynogenu) [3], [12].Zasada metody:Trawienie enzymatyczne białek prowadzi do powstania licznych produktów rozpadu. Wśród tych fragmentów znajdują się także krótkie peptydy, dla których określenie składu oraz właściwości możliwe jest jedynie po ich wyodrębnieniu z otrzymanej po trawieniu mieszaniny. Metoda izolacji krótkich peptydów zazwyczaj składa się z dwóch etapów:
- w pierwszym etapie za pomocą filtracji żelowej oddziela się małe peptydy od tych większych (fragmentów łańcuchów polipeptydowych trawionego białka).
- w drugim etapie za pomocą chromatografii fazy odwróconej możliwe jest frakcjonowanie peptydów wykorzystując w tym celu różnicę ich hydrofobowości [3], [12].
Wszystkie bufory i próbki stosowane w chromatografii HPLC muszą być filtrowane (filtry o porowatości od 0,45 do 0,6 µm) [3].
Trawienie fibrynogenu:Fibrynogen (o stęż. 10 mg/ml) należy poddać dializie wobec PBS w temperaturze 4st.C przez 3 godziny. Następnie, do próbki dodać plazminę – do końcowej aktywności 0,5U/ml. Prowadzić inkubację próbki w temp. 37st.C przez kolejne 3 h. Trawienie należy zastopować poprzez dodanie trasylolu (500 kU/ml). Uzyskany w ten sposób materiał zastosować następnie do filtracji żelowej [3], [12].
Filtracja żelowa hydrolizatu fibrynogenu:Należy odważyć 4,5g złoża Sephadex G-50 Fine, zalać wodą i pozostawić żel do napęcznienia (ok. 30 minut). Po upływie 30 min, zebrać supernatant znad napęczniałego żelu, po czym ponownie zalać go 20 ml wody. Odczekać ok. 10 min i zebrać supernatant wraz z zawiesiną bardzo drobnych fragmentów żelu. Czynność tę należy powtórzyć 3 razy, zamiast wody użyć bufor PBS. Przygotowane złoże upakować w plastikowej kolumnie o średnicy 1cm i długości 60 cm. Następnie, na kolumnę nanieść ok. 1 ml strawionego wcześniej fibrynogenu (nie więcej niż 3% objętości kolumny). Po wniknięciu próbki w złoże przez kolumnę przepuścić bufor PBS- przez cały czas należy zbierać 5 ml frakcje, w których za pomocą metody spektrofotometrycznej (przy λ=280nm) należy monitorować zawartość białka [3], [12].
Rozdział należy prowadzić aż do momentu przepuszczenia przez kolumnę ok. 500 ml buforu PBS (tj. 10-krotnej objętości kolumny). PO rozdziale należy narysować wykres zależności gęstości optycznej od objętości elucji (wykres chromatograficzny), na którym należy zaznaczyć ostatni jego szczyt. Wszystkie zebrane eluaty zawierające nisko cząsteczkowe peptydy połączyć razem, następnie rozdzielić na 1-mililitrowe frakcje (porcje) i zamrozić. Na koniec przez kolumnę chromatograficzną przepuścić 100 ml buforu PBS z dodatkiem 0,01% azydku sodu. Kolumnę przechowywać w temperaturze pokojowej - pilnując, by nie doprowadzić do wyschnięcia złoża [3], [12].
Rozdział chromatograficzny peptydówDo właściwego rozdziału peptydów należy stosować gradientowy system wysokosprawnej chromatografii cieczowej (HPLC). Kolumnę C18 zrównoważyć wodą z dodatkiem 0,1% TFA. Przepływ ustawić na 1 ml/min, z kolei detektor ustawić na λ=280 nm. Materiał (próbkę), którą ma isc do analizy należy przefiltrować przez filtr o średnicy porów równej 0,45 µm, następnie nanieśc 1 ml filtratu. Program chromatograficzny przygotować z liniowo narastającym gradientem acetonitrylu z dodatkiem 0,1% TFA w granicach 0-100% w czasie 20 minut. Narastanie stężenia acetontrylu uruchomić po ok. 5 minutach od momentu nastrzyknięcia próbki. Za pomocą kolektora należy zbierać 1-mililitrowe frakcje , a następnie na podstawie chromatogramu uzyskanego w integratorze zidentyfikować frakcje zawierające oczyszczone peptydy, po czym przeznaczyć je do dalszej analizy. Kolumnę po rozdziale zrównoważyć 20% roztworem acetonitrylu i przechowywać w temperaturze pokojowej [3], [12].
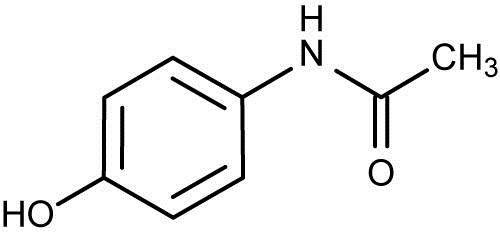
Oznaczanie leków w płynach fizjologicznych- metody analityczne [4], [12].Paracetamol (łac. Paracetamolum) – N-acetylo-p-aminofenol- należy do silnych leków przeciwbólowych . Zaliczany jest do pochodnych acetanilidu. W handlu paracetamol znajduje się od 1955 roku, z kolei w Polsce stał się popularny w latach 90. XX wieku, wypierając tym samym z rynku powszechnie znany i często używany lek przeciwgorączkowy znany jako piramidon [5]. Paracetamol ulega metabolizowaniu tj. nieuczynnieniu w wątrobie w wyniku 2 reakcji: sprzęgania oraz utleniania.
Na drodze sprzęgania paracetamolu z resztą kwasu siarkowego i glukuronowego powstają nieaktywne i nietoksyczne metabolity: siarczany (30%) i glukuroniany (50%). Związki te nie ulegają zwrotnemu wchłanianiu w kanalikach nerkowych przez co są wydalane wraz z mocze [6]. Około 5% paracetamolu zostaje wydalone w postaci niezmienionej przez nerki. W 5% do 20% dochodzi do metabolizmu zależnego od układu enzymatycznego cytochromu P450, a głównie przez jego izoformę znaną jako CYP2E1( rzadziej przez izoformy CYP1A2 i CYP3A4). Enzymatyczne utlenianie niewielkiej ilości paracetamolu (3%) przy udziale CYP2A6 i CYP2B1 prowadzi do powstania 3-hydroksy-paracetamolu, który po metylacji sprzęgany jest z wytworzeniem glukuronianów i siarczanów wydalanych w moczu [4], [6], [12].
Zdjęcie: Wzór paracetamolu, http://chemistry.about.com/od/factsstructures/ig/Chemical-Structures---A/Acetaminophen---Paracetamol.htm, ChemAxon.com/Marvin
W wyniku reakcji utleniania paracetamolu przy udziale izoformy CYP2E1, która jest obecna w nerkach, powstaje toksyczny metabolit :N-acetylo-4-benzochinonoiminy (w skrócie NAPQI). NAPQI jest bardzo silnym utleniaczem, który reaguje z grupami -SH w zredukowanym glutationie. To właśnie on odpowiedzialny jest za rozwój ewentualnego ciężkiego zatrucia paracetamolem. Do zatrucia dochodzi w przypadku wysycenia obu szlaków metabolicznych spowodowanych m.in. przekroczeniem dawek terapeutycznych lub wyczerpaniu ustrojowych zapasów siarczanów i glukuronianów [6], [7]. Dzieje się tak, ponieważ wysycenie szlaku metabolicznego powoduje wzmożone metabolizowanie paracetamolu na drodze utleniania i powstawania NAPQI.
NAPQI ulega detoksykacji poprzez nieenzymatyczną reakcj z grupami sulfhydrolowymi glutationu lub też z innymi związkami, które zawierają grupę tiolową (N-acetylocysteina), z ostatecznym wytworzeniem sprzężenia z kwasem merkapturowym. Powstałe w ten sposób nieaktywne i nietoksyczne metabolity są wydalane przez nerki [6], [7]. Wyczerpanie komórkowych rezerw glutationu i kumulacja NAPQI prowadzi do całkowitego zniszczenia wątroby i śmierci [8]. U niemowląt paracetamol jest metabolizowany głównie do siarczanów, z kolei u osób dorosłych paracetamol jest przede wszystkim sprzęgany z kwasem glukuronowym.
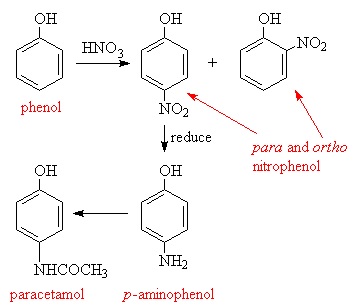
Zdjęcie: Otrzymywanie paracetamolu, http://www.ch.ic.ac.uk/rzepa/mim/drugs/html/paracet_text.htm
W przypadku, gdy spożycie paracetamolu przekroczy >10 g, dochodzi do wyczerpania glutationu z następczym toksycznym uszkodzeniem hepatocytów przez NAPQI, który wchodzi w reakcje z białkami. Wtedy też dochodzi do tworzenia się martwicy centralnej części zrazików – tj. obszaru charakteryzującego się największą aktywnością izoformy CYP2E1 i najmniejszym stężeniem glutationu [9]. Głównym miejscem toksycznego działania NAPQI jest wątroba, w mniejszym stopniu dochodzi też do uszkodzenia nerek, mięśnia sercowego oraz trzustki. Ponadto, wydostający się z hepatocytów NAPQI uszkadza również białka osocza, hemoglobinę i nabłonek płuc [6].
Paracetamol wchłaniany jest drogą jelitową,a następnie metabolizowany w wątrobie. W przypadku, gdy stężenie paracetamolu w osoczu przekroczy 2 mM dochodzi do poważnego uszkodzenia wątroby [12].
TeofilinaJest lekiem ułatwiającym oddychanie w chorobach somatycznych. Ponadto, stosuje się ją w leczeniu i zapobieganiu dychawicy oskrzelowej, a także przy nadciśnieniu i w chorobie wieńcowej [10]. Efekt terapeutyczny teofiliny skorelowany jest z jej stężeniem w osoczu, które powinno znajdować się w granicach 40 do 80 µM. Jak wykazały badanie, przekroczenie stężenia tego leku powyżej 100 µM powoduje wystąpienie licznych skutków ubocznych [Kłyszejko s. 176-177].
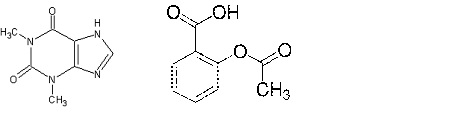
Zdjęcie: Teofilina–wzór(lewa strona) i kwas salicylowy –wzór (prawa strona) http://www.farmakognozja.farmacja.pl/alkaloid/index.php?alka=teofil, http://www.sigmaaldrich.com/catalog/product/sigma/a5376?lang=pl®ion=PL
Acetylosalicylany (tj. salicylany w formie eacetylowanje) są najczęściej stosowanymi leki przeciwbólowymi, przeciwzapalnymi, a także przecizakrzepowymi. Wchłaniane są drogą jelitową, zaś ich działanie polega na wiązaniu się z licznymi białkami osoczowymi. Stężenie salicylanów w osoczu nie powinno przekraczać stężenia 2,5 mM, ponieważ większe dawki mogą powodować poważne zaburzenia metaboliczne, a także wystąpienie lokalnych, niekontrolowanych krwawień [11], [12].
Autor: Lidia Koperwas
Literatura:
[1]. J. K. Rumiński "HPLC. Wysokosprawna chromatografia cieczowa", UMK, Toruń 2004
[2]. http://www.biofizyka.p.lodz.pl/ch7.pdf
[3]. Cierniewski C.S., Budzyński Z., 1993. Localization of the cross-linking site of GPRVVERHK in the ɣ-chain of human fibrinogen. Eur. J. Biochem., 218: 321 – 325
[4]. Wallinder H. High performance reversed phase chromatography in clinical chemistry: Separation of paracetamol, theophylline and salicylate in serum. Liquid chromatography. Application Note 368, LKB Bromma, Sweden.
[5].History of Tylenol (ang.). [dostęp 17 grudnia 2012].; http://www.nancywest.net/pdfs/McNeilConsumerHealthcareCompany.pdf
[6]. K.D. Rainsford: AspirinandRelatedDrugs. CRCPress, 2004.
[7]. Nonsteroidal Anti-inflammatory Drugs. W: William O. Foye, Thomas L. Lemke, David A. Williams: Principles of Medicinal Chemistry. Williams & Wilkins; 4th edition, 1995, s. 544–545. ISBN 0-683-03323-9.
[8].Grieb P., Prof. Dr. Hab. WPŁYW LEKÓW NA PROCESY WOLNORODNIKOWE W ORGANIZMIE. „FARMACJA POLSKA Czasopismo Polskiego Towarzystwa Farmaceutycznego”. Tom LVII, s. 01–15.08., 2001.
[9].Toxic responses of the liver. W: M Treinen-Moslen: Casarett and Doull's Toxicology. The basic science of poisons. Sixth Edition. McGraw-Hill Medical Publishing Division, 2001. ISBN 0-07-134721-6.
[10]. http://www.farmakognozja.farmacja.pl/alkaloid/index.php?alka=teofil
[11]. Andrew T. Chan, MD, MPH; JoAnn E. Manson, MD, DrPH; Diane Feskanich, ScD; Meir J. Stampfer, MD, DrPH; Graham A. Colditz, MD, DrPH; Charles S. Fuchs, MD, MPH. Long-term Aspirin Use and Mortality in Women. Arch Intern Med. 2007;167(6):562-572. doi:10.1001/archinte.167.6.562, http://archinte.jamanetwork.com/article.aspx?articleid=412018
[12]. Kłyszejko-Stefanowicz L, 2003. Ćwiczenia z biochemii. Wydawnictwo Naukowe PWN, 2003, s. 172 - 176
https://laboratoria.net/artykul/18625.html








Recenzje